
-
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Publish Your Research/Review Articles in our High Quality Journal for just USD $99*+Taxes( *T&C Apply)
Offer Ends On
Henry E. Young* and Oscar Tellez
Corresponding Author: Henry E. Young PhD, Chief Science Officer, Dragonfly Foundation for Research and Development, 12443 Venice Blvd (Corporate Office), Foley, AL 36535 USA.
Received: June 19, 2026 ; Revised: June 21, 2026 ; Accepted: June 22, 2026 ; Available Online: June 23, 2026
Citation: Young HE & Tellez O. (2026) Introduction to Adult Telomerase Positive Stem Cells (aTPSCs). J Stem Cell Ther Res, 1(1): 1-52.
Copyrights: ©2026 Young HE & Tellez O. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Views & Citations
Likes & Shares
The field of regenerative medicine has long sought the “Holy Grail”, a cell that has unlimited proliferation potential, can differentiate into any cell, can restore dead and dying cells to normal functional cells, and can be used for anyone, making them a universal regenerative stem cell. Many types of stem cells have been suggested to be the Holy Grail. Most notable are three categories of stem cells that have been widely studied since 1990. 1. Embryonic stem cells (ESCs) that are isolated from the inner cell mass of developing embryos, 2. Mesenchymal stem cells (MSCs) that were originally isolated from bone marrow of post-natal adults, and 3. Induced pluripotent stem cells (iPSCs) that are derived by transfecting embryonic genes, e.g., Oct-4, SOX2, c-Myc, and Klf4, into an adult differentiated cell, most notably adult dermal fibroblasts. Each one has advantages and disadvantages. Both ESCs and iPSCS, because of the presence of the telomerase enzyme have unlimited proliferation potential. Both ESCs and iPSCs are very plastic, they can differentiate into any somatic cell of the body. Unless prevented to do so though, both ESCs and iPSCs will form teratomas (cancerous cells) by spontaneous differentiation. To prevent teratoma formation, both ESCs and iPSCs need to be pre-differentiated, which in the process, loses their plasticity for forming multiple cells. The ESCs, by virtue of being isolated from the inner cell mass of developing embryos, are allogeneic, expressing self-recognition molecules which will induce a graft versus host disease (GvHD) response in the recipient. The iPSCs, being isolated from the same individual, were thought to negate the GvHD response. Unfortunately, the transfection process alters the self-recognition molecules to an extent to make them initiate a GvHD response. The MSCs have a limited lifespan of 70 population doublings before they senesce and die. MSCs, like all telomerase negative progenitor cells, decrease in number with increasing age of the individual. MSCs only form fat, cartilage, and bone, and therefore are not plastic in the ability to form all somatic cell types. Autologous (same person) MSCs do not elicit a GvHD, whereas (donor) allogeneic MSCs induce a GvHD due to presence of MHC Class-1 self-recognition cell surface molecules. In contrast to the above, we would like to offer a fourth category of stem cells for consideration, endogenous adult telomerase positive stem cells (aTPSCs). The aTPSCs retain the telomerase enzyme after birth that endows them unlimited proliferation potential. They are present throughout the lifespan of the individual. Collectively, they will form any cell of the conceptus, including all somatic cells of the body, gender-specific gametes, the nucleus pulposus of the intervertebral disc, and extraembryonic membranes, placenta and umbilical cord. Their default state is that of a dormant, quiescent, hibernating cell. They have to the stimulated by biological agents to do anything, hence, no teratoma formation because there is no spontaneous differentiation. They are very tightly controlled with respect to function: proliferation, progression, induction, and anti-differentiation.
Keywords: Adult, Telomerase Positive, Totipotent, Pluripotent, MSCs, ESCs, iPSCs
PODCAST - 1
If adult salamanders can regenerate tissues, why can’t humans?
Can you take us back to that moment?
Why did you ask that question, and why did it become important for your career?
Actually, the initial question for my Master’s thesis was: if juvenile salamanders can regenerate a limb, why can’t adult salamanders?
I read the existing literature on the subject at that time, circa 1950-1973 [1-12]. The multiple groups that had studied the phenomenon of limb regeneration in aquatic juvenile and adult newts and aquatic salamanders kept them at 4oC water, and fed beef liver daily during daylight hours. They assayed limb regeneration every five days for 30 days (newts) to 45 days (salamanders) for 6-9 time points. The investigators concluded that the juvenile aquatic salamanders damaged tissues dedifferentiated into the blastema and regenerated the limb [2].
They also concluded that limb regeneration did not occur in adult salamanders, which they kept under the same conditions and viewed them at the same time points.
From their observations they concluded that adult salamanders had lost the ability to regenerate a limb.
So, for my study, I kept the adult salamanders under the same environmental conditions as the juvenile aquatic species: 4oC water and fed beef liver daily during daylight hours. Using these environmental conditions, all my adult salamanders died of starvation before I could even start my experiments. So, I went to my Chairman and asked how I should proceed.
He said “what do you know of your model system?” – answer “they are adult salamanders”.With a twinkle in his eyes, he said “you need to dig deeper and find out everything you can about your model system”From my own field work observations and further literature research, I discovered that adult salamanders:
1. Are terrestrial, not aquatic, they hide in burrows in the ground during daylight hours.
2. They come out at night looking for a nocturnal (active at night) food source, preferably one that moves on its own.
3. Their preferred food source is cockroaches, although they were also preferential to night crawlers.
4. Migrate during the first cold rain of the fall, they spawn to the ponds they were born in to breed (copulate) with the opposite gender.
5. They did not eat when they were breeding.
6. When breeding was concluded, they return to their burrows
When I presented my findings to my Chairman, he told me four things that have followed me throughout my research career.
1. Know you model system
2. Tissue NEVER lies
3. You need to understand what your model system is telling you, and act accordingly
4. Just because something hasn’t been reported, does not mean it doesn’t exist, all it means is that it hasn’t been discovered yet.
So, I built a large, deep, terraria for all the salamanders, similar to environmental conditions in the wild. I released night crawlers into their terraria every two days. They apparently liked their conditions because weights increased. I repeated the experiment with fat, happy, and sassy salamanders. I kept the same observation times: every five days. But because it took the adult terrestrial salamanders over 370+ days to fully regenerate a limb, I was making 74+ observations, depending on the particular species in the genus Ambystoma (maculatum, annulatum, tigranum, and texanum).
And I discovered some interesting points (Figure 1):
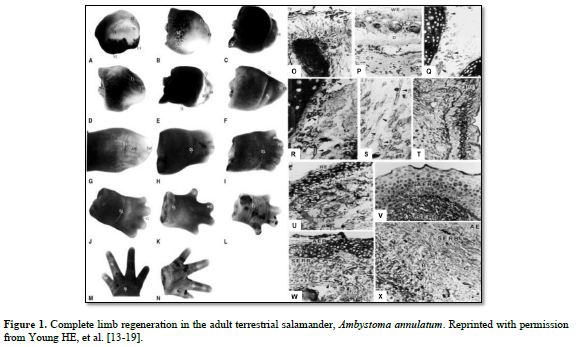
There was the same series of events that occurred in each of the four adult salamanders I examined, Ambystoma maculatum, annulatum, texanum, tigranum:
1. After amputation of the limb a transitional scar formed covering the wound site, basically a band-aid separating a very hostile external environment from a very delicate internal environment (O). Next, there was the appearance of macrophages that appeared underneath the transitional scar and cleaned out all the debris and dying cells, making the wound area sterile (P). There was formation of an apical epidermal ridge of non-descript cells on top of the transitional scar (B, C).
2. This ridge of cells began secreting a concentration gradient of sulfated, carboxylated, and neutral glycoproteins into the area through and underneath the transitional scar (U & V).
3. Previously very small unobserved cells, now covered in halos of heparan sulfate-PGs (HS-PG) (Q), broke loose from the more proximal connective tissues of the dermis; periosteum; perichondrium; muscle endomysium, perimysium, epimysium; nerve endoneurium, perineurium, and epineurium; and connective tissues surrounding the vasculature (R,S) and migrated to an area beneath the AER (T), eventually forming the sub-epidermal ridge blastema (SERB) (T- X)
4. The very small cells shed the HS-PG coverings and formed an indistinct mass of very small cells (T).
5. Then the very small cells they began to proliferate and physically push the AER outward (B-L).
6. This formed a gradient of differentiated tissues: differentiated tissues of the non-transected limb, through intermediaries similar to same tissues during embryonic development, to non-distinct cells of the “blastema”.
7. As the length of the appendage increased, the more proximal intermediaries turned into adult differentiated tissues.
Bottom line from those experiments:
Adult terrestrial salamanders regenerate perfectly fine, if given the appropriate environment, diet, exercise (hunting for food), and sleep cycle.
So, from knowledge of what occurred in adult salamanders spawned the question:
If adult salamanders can regenerate tissues, why can’t humans?
Or better yet, are we keeping humans under the wrong environmental conditions for regeneration to occur?
Why did you ask that question, and why did it become important for your career?
My parents and a close family member, had/have serious genetically inherited and acquired health issues. These health issues included heart disease and diabetes (father), Hashimoto’s disease, Systemic Lupus Erythematosus, Pulmonary Fibrosis, SLE-induced glaucoma, and SLE-induced Dementia (mother), and all of the above ‘inherited’ from both parents (close family member). Plus, he was diagnosed with Autoimmune Constellation Syndrome. To have this diagnosis one has to have a minimum of five autoimmune or autoimmune associated diseases. He has over 30. To give you an idea of what he had to deal with since he was 4 years old (in order of appearance): Hashimoto’s disease maintaining a short stature (4’5” tall) and overweight to severely obese (250-350 lbs.) from 4-17 years of age; Sjogren’s disease; Scleroderma; Alopecia; multiple allergies to foods, apparel, smoke, environment; adult respiratory distress syndrome (ARDS), pericarditis; pleuritis; pulmonary fibrosis; Rhinitis; Esophagitis; Tracheitis; Gastritis; Ileitis; Celiac Disease; Colitis; Rectifies (severe inflammation within the rectum); Hepatitis; Rhabdomyositis (severe inflammation of skeletal muscle), Rhabdomyolysis (wasting of skeletal muscle, think directed sarcopenia); Pancreatitis; Cholecystitis (Gall Bladder); Nephritis; Vasculitis; Systemic Lupus Erythematosus; osteopenia/osteoporosis (long term prednisone use), torsion (spiral) fracture of left leg into multiple pieces (due to a twisting fall); sterility; extreme sensitivity to sun light (photosensitivity) with resulting formation of keratoses; Neuropathies; Bi-Lateral Sciatica; Migraines; Cluster Headaches (Suicide Headaches); fibrosed CNs L1-S5 to his vertebral column (he said it felt like he was growing a dorsal fin from his vertebral column outward); extreme unrelenting pain (with the following pain killers given simultaneously every four hours: 64-mg of hydromorphone, 4x max dose Gabapentin, 2x max dose Baclofen, maxed out 12-hr Tylenol, Aspirin, Ibuprofen, and Naproxen, did not give him any relief from the pain); OIC (Opioid-induced constipation); TIAs (transient ischemic attacks, mini strokes); Cardiomyopathies; Tachycardia inducing Heart attacks, Autoimmune-induced Type-1 Diabetes; Chronic Kidney Disease; Rheumatoid Arthritis; Atrial Fibrillation; SLE-induced glaucoma; and cataracts. Suffice it to say, in the 70 years that he has been expressing various autoimmune and associated diseases, he states that it has been an interesting journey [20].
If these previously unrecognized primitive cells that I discovered in adult salamanders were also present in humans, could I restore the health of individuals in my family? That started my 50+ year quest.
2. Why are Salamanders so important?
Salamanders are the highest order of animal that will completely regenerate a limb that is an exact duplicate of the histoarchitecture of the limb that was lost.
What can they do that makes scientists ask bigger questions about healing and regeneration?
Scientists can ask about genetic control of regeneration; where are the genes that control the process; is epigenetics involved; is methylation involved; how are the biological clocks of various organs related to regeneration; are components of the ECM (extracellular matrix, e.g., collagens, proteoglycans, glycoproteins) involved in the process, and if so, how; will the cells involved spontaneously form a limb or is it a tightly controlled; are there biological factors involved that control the process; where are those factors located; how do they interface with the primitive cells; what are the characteristics of these cells; what techniques can you use to identify them; so on and so forth; and lastly, are similar scenarios and components present in humans. If so, how can they be used to restore damaged tissues in humans.
For example, for my PhD degree I performed glycoconjugate histochemistry on serial sections of the regenerating limb tissues to identify particular proteoglycans (PGs): chondroitin sulfate-PG, keratan sulfate-PG, dermatan sulfate-PG, chondroitin sulfate/keratan sulfate-PG (also called Aggrecan), non-sulfate chondroitin-PG, hyaluronic acid, sulfated glycoproteins, neutral glycoproteins, and carboxylated glycoproteins, and using microspectrophotometry, quantify their amounts. I also used scanning electron microscopy coupled with glycoconjugate histochemistry and X-ray energy dispersive microanalysis to quantity ECM components. What I discovered was that each tissue in the body, be it fully differentiated, newly forming, partially regenerated, or regenerated, had a unique glycoconjugate profile, which I called its “fingerprint”. From there, I wanted to be able to isolate these glycoconjugates in a biologically active form to see what effects they would have on the aTPSCs. After obtaining my PhD degree (1984), I obtained a postdoctoral fellowship in a laboratory that performed glycoconjugate biochemistry to isolate and characterize proteoglycans within the ECM [21-23].
3. Human Regeneration Question
What does regeneration mean to you?
Regeneration to me means restoration of the damaged and/or missing cells and tissues recreating the normal histoarchitecture of the lost tissues, and thereby restoring normal function.
4. Early Scientific Environment
When you began exploring this idea, what was the scientific field focused on?
Were people open to the idea that adult humans might still have still powerful cells, or was that idea outside the mainstream?
Basically, it was outside mainstream thought processes. This was because true adult stem cells had not yet been discovered. Therefore, common belief, based of dogma, said that these particular stem cells did not exist.
To be able to receive government funding to study any phenomenon, one of my PhD mentors told us that the game plan for receiving NIH funding was to do the experiments ahead of time, but wait to publish. Write the experiments performed as an application for a grant; including hypothesis, M&Ms, costs, etc. and submit. When that grant was funded, work on experiments for the next grant submission. At termination period of the first grant, publish the results. So, you would have fulfilled what you set out to prove, or disprove. In addition, it was far better to have a “story” to tell using multiple technologies, then to perfect a single technology and using the same technology on multiple tissues. So, my “story” for my research career has been the role of aTPSCs in regenerative medicine.
I isolated aTPSCs from chickens, cloned them from single cells using conditioned medium, characterized them, etc., and submitted the grant to NIH (circa 1989).
My “pink sheet” response from the grant reviewers was “well written grant, but your data is flawed. Everyone knows (dogma) that adult stem cells don’t exist. … But if they did exist, you would need to show them in a research animal which is preferably a mammal, not a chicken. We would suggest a mouse”.
I published the chicken methodology data: ELICA and Isolation protocols to a third-tier journal, Journal of Tissue Culture Methods. With respect to this particular journal, one would submit their manuscript to the editor of the journal. The editor would send your manuscript to one of the reviewers to repeat your experiments exactly as written. If they could not repeat your experiments and get exactly the same results, the manuscript was either rejected outright or revised significantly to match the permutations of your methodologies to get the experiments to work. Both manuscripts were accepted without revision [24,25].
I then started using mice: Balb-C (standard research mouse) and CBF-1 (NIH’s aging model) as my research subjects and repeated the experiments. The lifespan of a Balb-C mouse is 24 months (equivalent to about 60 years of age), whereas the lifespan of a CBF-1 mouse is 36-40 months (equivalent to about 120 years of age, the pre-programmed limit for humans) [21-23].
I joined Dr. Arnold Caplan’s lab in 1984 (nine years after I had discovered the aTPSCs in adult salamanders) for a postdoctoral fellowship in glycoconjugate biochemistry. Again, I wanted to isolate the glycoconjugates in their biological active form so I could apply them to the aTPSCs to determine if they were involved in cellular regeneration.
Being a biochemist by training, Dr. Caplan was a “lumper” with respect to anatomical structures. Being an anatomist/histologist/histochemist by training, I was a “splitter” with respect to anatomical structures. So, while Dr. Caplan viewed skeletal muscle as a single organ, I viewed skeletal muscle as a collection of individual tissues. There are three levels of structural elements composing skeletal muscle the organ. The first level is composed of mature myotubes having myosatellite cells (myoblast progenitor cells) outside their plasma membrane, but inside their basement membrane (consisting of type-IV collagen, entactin, nidogen, insoluble fibronectin, etc.), each myotube was surrounded by loose fibrous connective tissue (type-1 and type-12 collagens. Type-12 collagen is the bridge molecules between type-1 collagen and its associated GPs and PGs of the ECM) termed the endomysium. Embedded within the endomysium were capillaries, aTPSCs, hyaluronic acid with attached CS-PGs. Collections of these myotube structures were bundled together to form fascicles, the second level. The connective tissue surrounding the bundled fascicles were a moderately dense fibrous connective tissue (type-1 and -12 collagens) called the perimysium. Embedded within the perimysium are arterioles, venioles, small lymphatic vessels, motor end plates, sensory muscle spindles, nerve fibers, aTPSCs, and hyaluronic acid with attached CS-PGs. At the third level, bundles of myotubes came together to form skeletal muscle the organ, surrounded by a dense regular connective tissue covering termed the epimysium (type-1 and -12 collagens). Contained within the epimysium were muscular arteries, muscular veins, lymphatics, nerve fibers, Golgi tendon organs, aTPSCs, and hyaluronic acid with attached CS-PGs. The epimysium is continuous with tendons (connecting adjacent muscles to each other or connecting muscle to bones) [21-23].
I learned to isolate and characterize extracellular matrix PGs from Dr. David Carrino in Dr. Caplan’s lab [23] and glycoproteins from Dr. Masaki Yanagashita during a visit to Dr. Vince Hascall’s lab at NIH NIDR.
I submitted my next NIH grant (1990) dealing with aTPSCs in mice, Balb-C (normal) and CBF-1 (aging). It was denied, because Dogma says that adult stem cells do not exist. But if adult stem cells did exist, the reviewers stated that I would need to show them in a larger mammal, such as a rat.
I submitted manuscripts concerning aTPSCs in the chicken [24,25] and in both normal age mice and aged mouse models [21-23]to tier one journals: Cell, Nature, Science, PNAS (Proceedings of the National Academy of Science, USA). They were either rejected outright because of Dogma – adult stem cells do not exist; or stuck in review for over two years.
I had named the telomerase positive MesoSCs in that original paper “adult mesenchymal stem cells”, because they could form 37 separate and unique cell types within the embryonic mesodermal lineage (mesenchyme).
The manuscripts were finally released back to me after Dr. Arnold Caplan published his seminal “adult mesenchymal stem cell” paper (Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991; 9:641-650 [26]) showing the discovery of an adult stem cell isolated from bone marrow that would form 3 cell types: fat, cartilage, and bone. After his publication, my submitted manuscripts for chicken and mouse endogenous stem cells [24,25] were finally returned rejected for not being novel with respect to adult mesenchymal stem cells.
I finally published our work with chicken and mouse telomerase positive mesodermal stem cells (TP-MesoSCs), calling them “adult pluripotent mesenchymal stem cells” because of their ability to form 37 separate cell types [27-30]. I should have called them mesodermal stem cells, but I wanted to demonstrate a distinct difference between Caplan’s mesenchymal stem cells which would only three cell types (white fat, hyaline cartilage, and intramembranous bone) [31,32] and my telomerase positive MesoSCs, which would form 37 distinct cell types to several tier-2 journals [26-28]. Again, in some articles I originally called them pluripotent mesenchymal stem cells for their ability to form 37 different cell types within the mesodermal lineage, instead of just three cell types formed by Caplan’s (telomerase negative) tripotent progenitor MSCs, fat, cartilage, and bone [26,31,32].
Based on Caplan’s seminal adult MSC paper [26], adult stem cells were finally acknowledged to exist. But they had short comings. They had a defined lifespan of 70 population doublings before they senesced and died; they decreased in number with increasing age of the individual; and they would only form three cell types, e.g., (white) fat, (hyaline) cartilage, and (intramembranous) bone [26,30-32]. Around the same time as Caplan’s MSC publication [26], other “adult stem cell” papers were published: adult neural stem cells [33-39], adult hematopoietic stem cells [40-46], adult liver stem cells [47-52], adult pancreatic stem cells [53 -57], adult lung cells [58-63], etc. The organ specific adult stem cells correlated with Caplan’s MSCs, e.g., their lifespan conformed to Hayflick’s Limit of 70 population doublings from birth, they decreased with increasing age of the individual, and they only formed organ-specific cell types.
In 1998, Dr Thomson published on the derivation human embryonic stem cells from human blastocysts (ESCs). These ESCs were isolated from the inner cell mass of developing embryo and were pluripotent in that they could form any somatic cell of the body [64]. They were equivalent to the differentiation potential of the inner cell mass of the developing embryo (Figure. 2) [65].
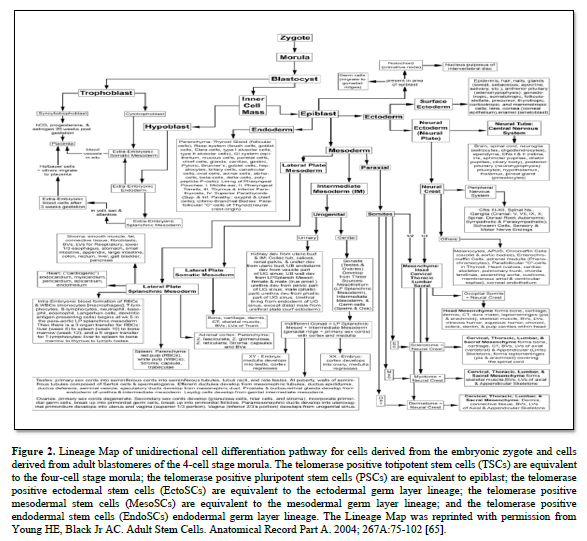
ESCs also contained the telomerase enzyme, which allows them unlimited proliferation potential [66-69]. Unfortunately, there were significant problems with ESCs when growing them outside the body (ex vivo) or transplanting them as naïve cells into the body. Unless they were physically prevented from differentiating with leukemia inhibitory factor (LIF) [70-77]; or stimulate to premature differentiation [78-82], ESCs will spontaneously differentiate into all somatic cells of an individual [83,84]. If the ESCs are in the uterus, they complete their normal pre-programming differentiation process forming an individual [84-86]. If they are outside the uterus, the ESCs will form a mass of somatic cells with no defined structure, called a teratoma cancer [87-91].
So now the politics. On one hand there were cell specific adult stem cells, that conformed to Hayflick’s Limit of 70 population doublings before they would senesce and die. They deceased with increasing age of the individual. Their differentiation was growth factor driven, but they weren’t as plastic as naïve ESCs [26,31,32,33-63].
On the other hand, ESCs would form every somatic cell of the body, proving to be very plastic. Containing the telomerase enzyme ESCs had essentially an unlimited proliferation potential. But, being allogeneic, they would express self-recognition markers of the donor and stimulate a GvHD, as well as forming teratomas if implanted in their naïve state [64-91].
So political debate was centered on ESCs versus MSCs, which one was better.
All the while we were publishing with collaborators in 2nd and 3rd tier journals on the aTPSCs [92-131]. As well as starting preclinical animal models of diseases: Parkinson’s disease [114], myocardial infarction [106], pulmonary fibrosis [107], and self-renewing immunoprotected pancreatic islet organoids for type-1 diabetes [110].
The political fall-out about using human embryos to derive embryonic stem cells lasted until Yamanaka (2009) published his seminal work on induced pluripotent stem cells (iPSCs) [132-134]. He placed embryonic genes (Oct-4, Sox2, c-Myc, and Klf4) into adult differentiated cells to mimic ESCs. And he did it so well with the transfection that the iPSCs expressed the same attributes as ESCs. The iPSCs expressed the telomerase enzyme [135-145], having an unlimited proliferation potential. In the naïve state, their inherent plasticity was their ability to form any somatic cell type in the body, which occurred spontaneously, just like ESCs [146-150]. This spontaneous differentiation occurred anywhere, in the culture dish, in an organism, etc., forming a teratoma (cancerous tissue) [146-150]. Unfortunately, to keep teratomas from forming they needed to pre-differentiate the cells into a single cell type [146-148]. By pre-differentiating the iPSCs or ESCs, they lose the naïve plasticity that made them a stem cell of choice for the Holy Grail.
5. What kept you curious?
You have spent decades studying this field. What kept you committed to this kind of research when most of the stem cell field was focused in other directions?
My early work with chickens, mice, and rats, demonstrated a very unique population of cells, with all the positives of ESCs and iPSCs, and “adult stem (progenitor cells)”, but none of the negatives:
1.Telomerase positive, so unlimited proliferationpotential as long as they stay uncommitted to aparticular lineage
2.Present throughout the lifespan of the individual
3.Found within connective tissue niches throughoutthe body
4.Will form literally any cell type in the body, e.g.,all somatic cells, gender-specific gametes, nucleuspulposus of intervertebral disc, and extraembryonicmembranes, placenta and umbilical cord
5.Proliferation is biological agent driven
6.Differentiation is biological agent driven
7.Anti-differentiation is biological agent driven
8.Once committed, progression is biological agentdriven
We have shown this same activity in 15 species of animals, including humans: amphibians (four species of adult terrestrial salamanders), reptiles (Komodo Dragon), avians (chickens and Wadel Crane), mice (Balb-c, CBF-1), rats (outbred Sprague-Dawley, inbred Wistar-Furth), rabbits, cats, dogs, sheep, goats, pigs, cows, bear (spectacled), horses, and humans (newborn to late geriatric) [151].
As I stated previously, this area is very personal to me. My family members had/have serious acquired and genetic health issues. If these previously unrecognized cells were present in humans, could I restore the health of my parents and myself, and in the process everyone else?
To achieve that goal, I like to think backwards (reverse chronological order) from my end goal, that gives me a straight-line pathway from start to finish:
End Goal: treating humans (and animals) with gender-matched universal aTPSCs world-wide.
24.Wide-spread treatment
23.FDA approval for commercialization
22.Testing CNSP vs Fresh isolate aTPSCs vs TSCs Ex vivo–determine safety & efficacy
21.Apply for IND from FDA for clinical trials
20.Clinical trials of Ex vivo propagated TP-TSCS – provesafety and efficacy
19.Apply for IND from FDA for clinical trials
18.Propagation of universal TP-TSCs Ex vivo
17.Clinical trials: Fresh isolate aTPSCs & CNSP to proveefficacy
16.Apply for IND from FDA for clinical trials
15.Treatment in humans (my family members).
14.Wide-spread IRB-approved clinical trials to prove safety(and efficacy)
13.Focused IRB-approved clinical trials to prove safety andefficacy
12.Pre-clinical animal models of disease
11.Characterization studies
10.Biobanking, Storage and Cryopreservation
9.Effects of biological agents on clones of aTPSCs
8.Generation of cell-specific exosomes,
7.Genomic labeling to track cells in vitro and in situ
6.Repetitive Single cell clonogenic analysis
5.Cell sorting
4.Cell surface marker profiles
3.Propagation
2.Plating
1.Isolation
Since FDA allows experimentation on oneself without reprimand, I was the first to receive an autologous transplant of aTPSCs (systemic delivery). My HIPPA code number is HM00001. My mind set at the time was, if the technology failed, I would be dead and the technology would not move forward.
My group started IRB approved compassionate use clinical trials in 2010. First in Parkinson patients, and then in COPD, IPF, and cardiomyopathy patients, matching the pre-clinical animal model systems [152-156].
I was also the first to receive a gender-matched, ABO blood group-matched allogeneic aTPSCs, by directed delivery and IV delivery. My mind set was the same, if the technology failed, I would be dead, and it would not move forward.
Positive results from the first trials allowed us to expand into other diseases: terminal, chronic diseases with no known cures, traumatic injuries, chronic orthopedic problems, autoimmune diseases, neurodegenerative, pulmonary, cardiovascular, and systemic [157-172].
It was too late to treat my father, because he passed away from a heart attack while I was still characterizing the cells. And it was too late to treat my mother, because she passed away from pulmonary fibrosis and dementia secondary to SLE while we were doing the preclinical animal studies. But I was just in time to treat my other family member. We had just started the IRB-approved compassionate use clinical trials for Parkinson’s disease and pulmonary diseases (COPD and IPF). I remember my PCP (board certified family physician) coming to me during a break in the phase tutorials, we both taught in the same phase. During break he put his arm around me and said “Henry, I know what you do for your research, go do it on yourself.” “Why?” “You have barely two weeks to live. You have already lost two organ systems and the remaining systems are operating at less than 25%. Your body is shutting down. You will be dead within two weeks if not sooner if you don’t save yourself.”
That night I discussed the situation with my wife. The next day I went to my chairman with letter in hand “As you know I have some serious health issues, I need about two weeks to go and get treated. If you don’t agree, here is my letter of resignation”. He said “Go for it and your job will be waiting for you when you return”.
And my wife did the same with her employer. “You know Henry is sick. He needs treatment or he will die. I need time off to take him to get treated. If you don’t agree, here is my letter of resignation”. They agreed as well.
In April of 2011 I had my first full autologous aTPSC transplant. Right after that first transplant, I was euphoric, absolutely no pain anywhere, I felt like Superman. The next day I woke up depressed, the extreme unrelenting pain was back. “Someone give me a gun I want to shoot myself”. The second day after treatment I woke up “Hey, this is strange, less pain than yesterday”. Third day same, less pain than day before. By the 7th day after treatment, I was neuropathically pain free, and basically have been ever since. But after a month, while there was no further downward progression of organ failure, I didn’t get any better. So, I had the first of nine allogeneic gender-matched, ABO-blood group-matched (3)and O-negative (6) aTPSC transplants. Those have beeninterspersed with 20 total autologous aTPSC transplants.With the allogeneic transplants, my signs and symptoms ofneurodegenerative diseases, cardiovascular morbidities,pulmonary fibrosis, chronic kidney disease, celiac disease,and SLE-associated morbidities began to reverse and myorgan functions began to increase. I topped out at levels thatwere 70% normal for a 20-year-old (acceptable to me).
My current mind set is, if my technologies, using either autologous and/or allogeneic aTPSCs, can bring my family member back from my death bed and give him a reasonable quality of life, then the aTPSCs should help people with other health problems as well.
Part 2. The Stem Cell Categories Most People Know
6.The Three Main Categories
At a high level can you explain what those three categories are:
ESCs – embryonic stem cells are derived from inner cell mass of embryo, they are pluripotent in their ability to form all somatic cell types of the body, they contain the telomerase enzyme for essentially unlimited proliferation potential. Initially published for humans in 1998 by Dr James Thomson [64].
iPSCs – induced pluripotent stem cells were generated by taking differentiated adult cells and transfecting into their nucleus four embryonic genes (Oct-4, SOX2, c-Myc, and Klf4) to have them mimic embryonic stem cells: pluripotent in ability to form all somatic cell types of the body, contains telomerase enzyme for essentially unlimited proliferation potential. Published by Yamanaka in 2009 [132].
MSCs – an “adult stem cell” (actually a tripotent progenitor cell) originally derived from bone marrow that will form three differentiated cell types: fat, cartilage, and bone. MSCs are telomerase negative. They have a lifespan of 70 population doublings before they senesce and die. MSCs decrease with increasing age of the individual. Published by Arnold Caplan in 1991 [26].
7.Embryonic Stem Cells
What makes embryonic stem cells so important clinically?
Embryonic stem cells were originally designed to study embryogenesis in utero: discovering genes and teratogens impacting signaling pathways, differentiation steps, etc., to determine how one could repair, for example, inborn errors of metabolism, spina bifida, Chiari syndrome, microcephaly, autism, cleft lip, cleft palate, etc., etc., etc., before the baby was born.
Then someone had the “bright idea” that they could use ESCs in adults (post-natal individuals) to repair acquired and genetic diseases.
What limitations have made them difficult to use clinically?
1.First, and foremost, is Politics – “killing an embryoto acquire ESCs. Embryos have rights too”.
2.Obtaining funding from the government because ofthe above to study ESCs.
3.Their spontaneous differentiation into multiple celltypes, necessitates using an inhibitory agent (e.g.,LIF) to prevent spontaneous differentiation.
4.ESCs formation of teratomas when transplanted invivo in a naïve state.
5.ESCs needed to be pre-differentiated beforetransplant to prevent teratoma formation.
6.ESCs are allogeneic (non-self).
7.ESCs express self-recognition cell surfacemolecules that will induce a graft versus hostdisease response in the recipient, HLA-DR markersfor hematopoietic lineage markers and MHC Class-1 markers for somatic cells that were not in thehematopoietic lineage.
8.Induced Pluripotent Stem cells
Can you explain what iPSCs are in simple terms?
The induced pluripotent stem cells were generated by taking differentiated adult cells, originally dermal fibroblasts/fibrocytes, but other cell types have been used as well. And using adenoviruses, transfecting four embryonic genes (Oct-4, SOX2, c-Myc, and Klf4) into the nucleus of the adult differentiated cells, to have them mimic embryonic stem cells. After which, they were pluripotent in ability to form all somatic cell types of the body, and expressed the telomerase enzyme for essentially unlimited proliferation potential.
Why was that discovery such a big deal?
iPSCs gave scientists a method to reprogrammed cells to a less differentiated cell type, e.g., a pluripotent cell that would form all somatic cells of the body. Since they were from the person’s own body the self-recognition cell surface markers would be the same so there would be no graft versus host disease (GvHD) response (theory).
9.Limits of Reprogramming
What are some of the challenges the field still has to solve?
Are there issues around safety, consistency, tumor risk, or clinical practicality?
Several of the major problems of iPSCs is that they mimic ESCs too well and have demonstrated the same inherent problems:
1.They form teratomas when transplanted in vivoin a naïve state.
2.Since they spontaneously differentiate intomultiple cell types, this necessitates using aninhibitory agent (LIF or some facsimile) to prevent spontaneous differentiation.
3.They need to be pre-differentiated to preventtumor formation.
4.And even though they come from the sameindividual, the reprogramming changes theexpression of the self-recognition molecules on their cell surfaces making them seem allogeneic to the recipient’s immune system, which will induce a graft versus host disease response in the recipient, destroying the iPSCs.
5.Labs are propagating the iPSCs at a doubling ratefaster than their cell cycle rate to increasenumber of cells generated. Unfortunately, as the doubling rate increases the number of mutations formed increases, and begins to increase exponentially at 10^9 cells.
6.Permanently mutated cells can have deleteriouseffects downstream in the treatment phase.
7.Only correct non-mutated iPSCs need/should tobe selected for human treatments.
8.It takes about 6-12 months to isolate, propagate,induce, select, and generate sufficient numbersof specific iPSC cell types for transplant. Increasing costs with respect to time, reagents, etc.
9.And lastly, from my own observations andresearch: the “body” does not likedifferentiated cell types, it views them as foreign, even those expressing the same MHC Class-1 markers. The body will wall them off from the rest of itself and encapsulates it with scar tissue. It prefers an undifferentiated cell that it can manipulate and dictate what it becomes.
10.Mesenchymal Stem Cells
MSCs have become the most talked about cell types in regenerative medicine.
What are MSCs?
And what do you think they can and cannot do?
Since previous to Caplan’s publication in the journal Science, adult stem cells were thought NOT to exist (Dogma). Now, here is a paper from a known scientist (biochemist) that says that ADULT STEM CELLS do exist in the form of mesenchymal stem cells. And that these adult stem cells can be isolated from adult bone marrow, and will form fat, cartilage, and bone.
Through my early years of my research, when I characterized aTPSCs (TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs), I also characterized the tripotent MSC, Caplan’s MSC. I characterized mixed isolates of aTPSCs and MSCs, clones of all six cell types derived from single cells derived by repetitive single cell clonogenic analyses, and from genomically-labeled aTPSCs clones compared to the unlabeled clone of MSCs. In this last instance, I sent the clones of aTPSCs and MSCs to Cecille Duplaa at INSERM in France to genomically label the cells. She tried to transfect all the clones with the Lac-Z gene for beta-galactosidase, but instead of using adenoviruses [scientifically accepted method, but sometimes the viruses can go “rogue” especially in long term culture of cells], she used lipofectin. Lipofectin “transfects” the cells during cell division. The more the cells divide during a given time frame the higher percentage of cells are transfected. The transfection rate for TSCs were 99%; for PSCs 98%; for EctoSCs, MesoSCs, and EndoSCs, greater than 95%; and for MS <5%.
With Arnold Caplan being the “lumper” that he was, and me being the “splitter” that I am as well as being a trained histologist/glycoconjugate histochemist/immunocytochemist, I also characterized the cell fates of his tripotent progenitor MSC, e.g., “fat (white fat), cartilage (hyaline cartilage), bone (intramembranous bone)”, using morphological, histochemical, and immunocytochemical criteria [130,131,151,173-175].
Table 1. Antibodies, Immunocytochemistry, & Histochemistry for Phenotypic Expression Markers
| Antibody | Antigen | Embryological Origin |
| CEA-CAM-1 | Carcinoembryonic antigen-cell adhesion molecule-1 | Totipotent |
| HCEA | Human Carcinoembryonic antigen | Totipotent |
| CEA | Carcinoembryonic antigen | Totipotent |
| CD66e | Carcinoembryonic antigen | Totipotent |
| DH-TuAg1 | Spermatogonia | Totipotent Gamete |
| MC-480 | SSEA-1 | Pluripotent |
| MC-631 | SSEA-3 | Pluripotent |
| MC-813 | SSEA-4 | Pluripotent |
| CD10 | Neutral endopeptidase | Pluripotent |
| AlkPhos | Alkaline Phosphatase | Pluripotent |
| CD56 | Neural cell adhesion molecule | Ectoderm |
| Pax-6 | Neurogenic lineage | Ectoderm |
| FORSE-1 | Neuronal precursor cells | Ectoderm |
| Vimentin | Cells of neurogenic lineage | Ectoderm |
| Nestin | Cells of neurogenic lineage | Ectoderm |
| R401 | Nestin-neuronal lineage | Ectoderm |
| HNES | Nestin-neuronal lineage | Ectoderm |
| MAB353 | Nestin-neuronal lineage | Ectoderm |
| RT-97 | Neurofilaments = neurons | Ectoderm |
| NF68 | Neurofilament-68 = neurons | Ectoderm |
| S-100 | Neurofilaments-100 = neurons | Ectoderm |
| NF-145 | Neurofilaments-145 = neurons | Ectoderm |
| N-200 | Neurofilaments-200 = neurons | Ectoderm |
| 8A2 | Neurons | Ectoderm |
| NG2 | Neurons | Ectoderm |
| TH | Tyrosine hydroxylase, precursor to neural transmitters | Ectoderm |
| SV2 | Synaptic vesicles | Ectoderm |
| DOPA | Dopamine, transmitter of dopaminergic neurons | Ectoderm |
| T8660 | Beta-tubulin-III | Ectoderm |
| Tuj1 | Beta-tubulin | Ectoderm |
| GFAP | Glial-fibrillary acidic protein | Ectoderm |
| CNPase | Glial cells = oligodendrocytes & astrocytes | Ectoderm |
| Rip | Oligodendrocytes | Ectoderm |
| MOSP | Oligodendrocyte specific proteins | Ectoderm |
| MAB | Oligodendrocyte marker | Ectoderm |
| 40E-C | Radial cells and radial glial cells | Ectoderm |
| VM-1 | Keratinocytes | Ectoderm |
| M3F7 | Type-IV collagen, basement membrane | Ectoderm & Mesoderm |
| 31-2 | Laminin, basement membrane | Ectoderm & Mesoderm |
| 5D2-27 | Cell adhesion molecule | Ectoderm & Mesoderm |
| B3/D6 | Fibronectin, basement membrane | Ectoderm & Mesoderm |
| 5C6 | Type-IV collagen, basemen membrane | Ectoderm & Mesoderm |
| Anti-type IV | Type-IV collagen | Ectoderm & Mesoderm |
| 33-2 | Heparan sulfate proteoglycan | Ectoderm & Mesoderm |
| Anti-HSPG | Heparan Sulfate proteoglycan | Ectoderm & Mesoderm |
| 5D4 | Keratan sulfate proteoglycan | Ectoderm & Mesoderm |
| 2E8 | Laminin, basement membrane | Ectoderm & Mesoderm |
| D3 | Desmin, in all 3 muscle groups | Ectoderm & Mesoderm |
| Anti-vimentin | Vimentin, lens of the eye | Ectoderm & Mesoderm |
| D76 | Desmin, in all 3 muscle groups | Ectoderm & Mesoderm |
| CD13 | Amino endopeptidase | Mesoderm |
| 12/101 | Skeletal Muscle | Mesoderm |
| C3/1 | Glycoprotein of myoblast plasma membrane | Mesoderm |
| OP-137 | MyoD | Mesoderm |
| F5D | Myogenin = skeletal muscle | Mesoderm |
| ALD-66 | Slow twitch muscle fibers | Mesoderm |
| MF-1 | Fast twitch muscle fibers | Mesoderm |
| MF-5 | Myosin light chain-2 of fast muscle | Mesoderm |
| MF-20 | Sarcomeric myosin = skeletal muscle | Mesoderm |
| MF-30 | Neonatal and adult myosin | Mesoderm |
| ALD58 | Myosin heavy chain | Mesoderm |
| CH1 | Myosin tropomyosin | Mesoderm |
| A4.74 | Myosin fast chain | Mesoderm |
| JLA-20 | Actin | Mesoderm |
| Anti-Myosin | Skeletal muscle myosin | Mesoderm |
| IA4 | Smooth muscle alpha actin = smooth muscle | Mesoderm |
| Calp | Calponin | Mesoderm |
| MAB-3252 | Cardiotin = cardiac myocytes | Mesoderm |
| MAB1548 | Myosin heavy chain of cardiac muscle | Mesoderm |
| M-38 | Type 1 collagen | Mesoderm |
| SP1.D8 | Procollagen type-III | Mesoderm |
| Anti-type-II | Type-II collagen | Mesoderm |
| WV1D1 | Bone sialoprotein II = bone | Mesoderm |
| Anti-OsteC | Osteocalcin / Bone Gla-protein | Mesoderm |
| MP111 | Osteopontine = bone | Mesoderm |
| Von Kossa | Stain calcium in bone | Mesoderm |
| EGTA | Leaches Calcium from bone, negative control | Mesoderm |
| CIIC1 | Type-II collagen | Mesoderm |
| II-4CII | Type-II collagen | Mesoderm |
| Anti-type2 | Type-II collagen | Mesoderm |
| HC-II | Human type-II collagen | Mesoderm |
| D1-9 | Type-IX collagen = cartilage | Mesoderm |
| 9/30 | Cartilage link protein | Mesoderm |
| 12/21 | Cartilage proteoglycan hyaluronate binding region | Mesoderm |
| 12C5 | Versican hyaluronate binding region | Mesoderm |
| H-DC34 | Sialomucin-containing hematopoietic/endothelial cells | Mesoderm |
| CD31 | PECAM, Peripheral endothelial cell adhesion molecule | Mesoderm |
| P1H12 | Human endothelial cell surface marker | Mesoderm |
| P2B1 | Peripheral endothelial adhesion molecule | Mesoderm |
| P8B1 | VCAM, vascular cell adhesion molecule | Mesoderm |
| P2H3 | CD62e, E-selectin (vasculature) | Mesoderm |
| H-endo | CD146, Endothelial cells | Mesoderm |
| H5A4 | CD11b, granulocytes, monocytes, NK-cells | Mesoderm |
| H4C4 | CD44, hyaluronate receptor | Mesoderm |
| Hermes-1 | CD44, hyaluronate receptor | Mesoderm |
| H5A5 | CD45, all hematopoietic cells except RBCs | Mesoderm |
| H5C6 | CD63, macrophages, monocytes, platelets | Mesoderm |
| HFSP | Human fibroblast specific protein | Mesoderm |
| 1B10 | Fibroblast-specific protein | Mesoderm |
| Sudan Black-B | Stains fat (adipocytes) | Mesoderm |
| Oil Red-O | Stains fat (adipocytes) | Mesoderm |
| H-AFP | Human alpha-fetoprotein = fetal liver | Endoderm |
| R-AFP | Rat alpha-fetoprotein = fetal liver | Endoderm |
| DESMO | Endodermal epithelial marker of liver | Endoderm |
| LAP | Canalicular cell surface protein of liver | Endoderm |
| 151-Ig | Liver epithelial growth factor | Endoderm |
| HA4c19 | Bile canalicular cells of liver | Endoderm |
| OC2 | Progenitor cells, oval cell, & biliary cells of liver | Endoderm |
| OC3 | Progenitor cells & biliary cells of liver | Endoderm |
| OC4 | Progenitor cells & biliary cells of liver | Endoderm |
| OC5 | Progenitor cells & biliary cells of liver | Endoderm |
| OC10 | Progenitor cells & biliary cells of liver | Endoderm |
| H.4 | Intracellular staining of liver hepatocytes | Endoderm |
| H.1 | Liver hepatocytes cell surface marker | Endoderm |
| DPPIV | Progenitor, canalicular, and biliary cells of the liver | Endoderm |
| OV6 | Biliary and oval cells of liver; biliary cells of liver | Endoderm |
| HESA | Human GI (Gastrointestinal) Epithelium | Endoderm |
| YM-PS087 | Glucagon-secreting cells of endocrine pancreas | Endoderm |
| YM-PS5088 | Insulin-secreting cells of endocrine pancreas | Endoderm |
| 11180 | Somatostatin-secreting cells of the endocrine pancreas | Endoderm |
| CK-19 | Ductal cells of the exocrine pancreas | Endoderm |
| ABL-93 | Lysosomal membrane glycoprotein | Ectoderm, Mesoderm, Endoderm |
| 22/18 | Regeneration cells | Ectoderm, Mesoderm, Endoderm |
| Telom | Telomerase positive cells | aTPSCs |
| CD90 | Glycosylphosphatidylinositol anchoring membrane protein (Thy-1) | Transition: EctoSCs, MesoSCs, and EndoSCs to Progenitor Cells |
| Thy-1 | Glycosylphosphatidylinositol anchoring membrane protein (CD90) | Transition: EctoSCs, MesoSCs, and EndoSCs to Progenitor Cells(e.g., MSCs) |
| CD95 | Cells undergoing apoptosis | Dead Cells |
| PI | Propidium Iodide, measure of live cells, flow cytometry | Live Cells |
| DAPI | Fluorescent marker to visualize living and fixed DNA | Live & Dead Cells |
| Gal-19 | Insect beta-galactosidase genomic marker | Cell tracking marker |
| Mallory Heidenhain One-Step | Identifies various cell types by color:Type-1 collagen – dark blueType-2 collagen – light blueSkeletal muscle – dark magentaCardiac muscle – intermediate magentaSmooth muscle – light magentaAdipose Tissue - whiteNerve fibers – lavenderRBCs - golden | Cells & Extracellular Matrix |
| Alcian Blue | Stains anions on carbohydrate & sulfate groups | Extracellular Matrix |
| AB 1.0 | Alcian Blue, pH 1.0 stains sulfate groups on GAGs | Extracellular Matrix |
| AB 2.5 | Alcian Blue, pH 2.5 stains carboxyl groups on GAGs | Extracellular Matrix |
| Alcec Blue | Stains anions on carbohydrate groups | Extracellular Matrix |
| AcB 1.0 | Alcec Blue, pH 1.0 stains sulfate groups on GAGs | Extracellular Matrix |
| AcB 2.5 | Alcec Blue, pH 2.5 stains carboxyl groups on GAGs | Extracellular Matrix |
| Safranin-O | Stains anions on carbohydrate & sulfate groups | Extracellular Matrix |
| SO 1.0 | Safranin-O, pH 1.0 stains sulfate groups on GAGs | Extracellular Matrix |
| SO 2.5 | Safranin-O, pH 2.5 stains carboxyl groups on GAGs | Extracellular Matrix |
| Enzyme | Streptomyces Hyaluronidase, negative staining control to verify presence of hyaluronic acid | Extracellular Matrix |
| Enzyme | Chondroitinase-AC, negative staining control to verify presence of chondroitin sulfate proteoglycans | Extracellular Matrix |
| Enzyme | Chondroitinase-ABC, negative control to verify presence of chondroitin non-sulfated proteoglycans | Extracellular Matrix |
| Enzyme | Keratanase, negative control to verify presence of keratan sulfate proteoglycans | Extracellular Matrix |
| Enzyme | Heparanase, to verify presence of heparan sulfate proteoglycans | Basement Membranes |
| PAS | Periodic Acid Schiff reaction for glycoproteins with vicinal hydroxyl groups | Extracellular Matrix |
Table 1. Immunocytochemistry with antibodies for cell-specific phenotypic expression markers and glycoconjugate histochemistry to determine “fingerprints” of specific cell types. Reprinted with permission from Young HE. A high throughput screening assay to quantify, visualize, and standardize biological activities: Enzyme-Linked Immuno-Culture Assay (ELICA). GSC Advanced Research and Reviews. 2025; 24(02): 091-114 [173]; Young HE, Speight MO. Osteoarthritis Treated with Telomerase-Positive Adult Stem Cells in Animals and Humans. Stem Cells Regen Med. 2020; 4(2):1-11 [158].
1. MSCs, specifically, form unilocular white fat. There are two types of fat: unilocular white fat and multilocular brown fat.
a. White fat
i. A single large vesicle is present filling the cytoplasm
ii. It has a single laterally-located nucleus
1. Lipid within the vesicle stains with oil-loving dyes, such as Oil Red-O and Sudan Black-B
b. Multilocular Brown
i. Is multilocular, having a centrally-located nucleus
ii. Multiple small vesicles are contained within the cytoplasm
iii. Lipid within the vesicle(s) stains with oil-loving dyes, such as Oil Red-O and Sudan Black-B.
2. MSCs, specifically, will form hyaline cartilage. There are five types of cartilage in the body:
a. Fibrocartilage
i. Appearance: Herringbone pattern of parallel dense regular connective tissue composed of collagen fibers, chondrocyte present within large oval-shaped lacunae
1. Located in symphysis pubis, menisci, labrum, and annulus fibrosis of intervertebral disc
ii. Collagen fibers are type-1 and type-12 collagen (bridge molecule)
1. Antibodies: M-38,
2. Mallory Heidenhain One Step – dark blue
3. Pure chondroitin sulfate proteoglycans
4. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
iii. Hyaluronic acid
1. Antibodies: H4C4 (CD44), Hermes-1 (CD44), 12C5 (HA binding region)
2. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Streptomyces hyaluronidase
b. Growth Plate Cartilage:
i. Location: metaphyseal portion of developing long bones during endochondral ossification and in hard callus during fracture repair
ii. Appearance: spicules composed of cartilage cores covered with lamellar bone, chondrocytes present in large oval lacunae, osteocytes present in small irregularly shaped lacunae
iii. Inner Cartilage cores
1. Collagens type-2 and type-9 (bridge molecule)
a. Antibodies: CIIC1, II-4CII, Anti-type2, Anti-type-II, HC-II,D1-9, 9/30, 12/21, 12C5
b. Mallory Heidenhain One Step: - light blue
2. Chondroitin sulfate/keratan sulfate proteoglycans (Aggrecan)
a. Glycoconjugate Histochemistry:
i. Chondroitin Sulfate Glycosaminoglycan chains
1. Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
ii. Keratan Sulfate Glycosaminoglycan chains
1. Alcian Blue pH 1.0, Alcec Blue pH 1.0, Safranin-O pH 1.0 with and without Keratanase
3. Hyaluronic acid
a. Antibodies: H4C4 (CD44), Hermes-1 (CD44), 12C5 (HA binding region)
b. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Streptomyces hyaluronidase
iv. Outer layers of Lamellar bone
1. Collagen fibers are type-1 and type-12 collagen (bridge molecule)
a. Antibodies: M-38,
b. Mallory Heidenhain One Step – dark blue
c. Pure chondroitin sulfate proteoglycans
d. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
2. Hyaluronic acid
a. Antibodies: H4C4 (CD44), Hermes-1 (CD44), 12C5 (HA binding region)
b. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Streptomyces hyaluronidase
c. Hyaline cartilage,
i. Location: attachment of ribs to sternum
ii. Appearance: random pattern of chondrocytes within large oval lacunae embedded with an amorphous extracellular matrix
iii. Extracellular Matrix
1. Collagens type-2 and type-9 (bridge molecule)
a. Antibodies: CIIC1, II-4CII, Anti-type2, Anti-type-II, HC-II,D1-9, 9/30, 12/21, 12C5
b. Mallory Heidenhain One Step: - light blue
2. Chondroitin sulfate/keratan sulfate proteoglycans (Aggrecan)
a. Glycoconjugate Histochemistry:
i. Chondroitin Sulfate Glycosaminoglycan chains
1. Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
ii. Keratan Sulfate Glycosaminoglycan chains
1. Alcian Blue pH 1.0, Alcec Blue pH 1.0, Safranin-O pH 1.0 with and without Keratanase
3. Hyaluronic acid
a. Antibodies: H4C4 (CD44), Hermes-1 (CD44), 12C5 (HA binding region)
b. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Streptomyces hyaluronidase
d. Elastic cartilage,
i. Location: Pinna of ear, eustachian tube, epiglottis
ii. Appearance: random pattern of chondrocytes within large oval lacunae embedded with an amorphous extracellular matrix containing elastic fibers
iii. Extracellular Matrix
1. Elastic fibers – elastin stain
2. Collagens type-2 and type-9 (bridge molecule)
a. Antibodies: CIIC1, II-4CII, Anti-type2, Anti-type-II, HC-II, D1-9, 9/30, 12/21, 12C5
b. Mallory Heidenhain One Step: - light blue
3. Chondroitin sulfate/keratan sulfate proteoglycans (Aggrecan)
a. Glycoconjugate Histochemistry:
i. Chondroitin Sulfate Glycosaminoglycan chains
1. Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
ii. Keratan Sulfate Glycosaminoglycan chains
1. Alcian Blue pH 1.0, Alcec Blue pH 1.0, Safranin-O pH 1.0 with and without Keratanase
4. Hyaluronic acid
a. Antibodies: H4C4 (CD44), Hermes-1 (CD44), 12C5 (HA binding region)
b. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Streptomyces hyaluronidase
e. Articular cartilage
i. Location: covering surface of articulating bones
ii. Appearance: 5 zones
1. Tangential zone
a. Type-2 and type-9 collagen fibers run parallel to surface,
b. Collagen fibers attach to lubricin,
i. A highly sulfated glycoprotein,
ii. forms a dipole with water,
iii. provides lubrication for articular joint
c. ECM consists of keratan sulfate proteoglycans only
2. Transitional zone
a. Type-2 and type-9 collagen fibers form crisscross pattern of fibers,
b. ECM consists of chondroitin sulfate proteoglycans only
3. Radial zone
a. Type-2 and type-9 collagen fibers run perpendicular to surface,
b. ECM consists of chondroitin sulfate/keratan sulfate proteoglycans attached to hyaluronic acid through a link protein (Aggrecan)
4. Tidewater Mark
a. Acellular
b. ECM consists of a jumbled mix of type-2 and type-9 collagen fibers, and a mix of chondroitin sulfate proteoglycans, keratan sulfate proteoglycans, and Aggrecan = chondroitin sulfate/keratan sulfate proteoglycans attached to hyaluronic acid through a link protein
5. Calcified cartilage
a. Chondrocytes present in intermediate-sized oval lacunae.
b. Type-2, type-9, type-1 and type-12 collagen fibers in a haphazard arrangement
c. ECM consists of Aggrecan = chondroitin sulfate/keratan sulfate proteoglycans attached to hyaluronic acid through a link protein, and calcium
d. Amorphous calcium phosphate
iii. Extracellular Matrix
1. Type-2 and type-9 (bridge molecule)
a. Antibodies: CIIC1, II-4CII, Anti-type2, Anti-type-II, HC-II, D1-9, 9/30, 12/21, 12C5
b. Mallory Heidenhain One Step: - light blue
2. Type-1 and type-12 collagen (bridge molecule)
a. Antibodies: M-38,
b. Mallory Heidenhain One Step – dark blue
3. Pure chondroitin sulfate proteoglycans
a. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
4. Pure keratan sulfate proteoglycans
a. Alcian Blue pH 1.0, Alcec Blue 1.0, Safranin-O pH 1.0 with and without Keratanase
5. Chondroitin sulfate/keratan sulfate proteoglycans (Aggrecan) double stained: first at pH 1.0 (Safranin-O) followed by pH 2.5 (Alcian blue or Alcec blue)
a. Chondroitin Sulfate Glycosaminoglycan chains
i. Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Chondroitinase-AC
b. Keratan Sulfate Glycosaminoglycan chains
i. Alcian Blue pH 1.0, Alcec Blue pH 1.0, Safranin-O pH 1.0 with and without Keratanase
c. Chondroitin sulfate/Keratan sulfate proteoglycan (Aggrecan)
i. Safranin-O at pH 1.0 followed by Alcian Blue (or Alcec Blue) at pH 2.5 with and without combined Chondroitinase-AC and Keratanase
d. Hyaluronic acid
i. Antibodies: H4C4 (CD44), Hermes-1 (CD44), 12C5 (HA binding region)
ii. Glycoconjugate Histochemistry: Alcian Blue pH 2.5, Alcec Blue 2.5, Safranin-O pH 2.5 with and without Streptomyces hyaluronidase
e. Amorphous calcium
i. Von Kossa stain with and without ethylene-glycine tetraacetic acid, a specific chelator for calcium
3. MSCs, specifically, will form intramembranous bone. During embryogenesis, bone forms by two mechanisms:
a. Endochondral ossification: mesodermal to cartilage model to bone, ex.; long bones, bones of face
i. Mesodermal cells
ii. Cartilage Model = growth plate cartilage (see above)
iii. Lamellar Bone
1. Heidenhain staining: Dark blue
2. Antibodies: M-38
3. Collagens: type-1 and type-12
4. Proteoglycans: Chondroitin sulfate-PG
b. Intramembranous ossification [direct mesoderm to bone]. ex., flat bones found in the calvaria, and flat portion of scapula.
i. Mesodermal cells
1. Lamellar Bone
2. Heidenhain staining: Dark blue
3. Antibodies: M-38
4. Collagens: type-1 and type-12
5. Proteoglycans: Chondroitin sulfate-PG
4. The first reported MSCs were a tripotent progenitor cell forming three cell types, white fat, hyaline cartilage, and intramembranous bone.
5. They are absent the telomerase enzyme.
6. They have a lifespan of 70 population doublings before they senesce and die.
7. They decrease in number with increasing age of the individual
8. The tripotent progenitor MSCs express CD90, CD105, CD123, CD166, and MHC Class-1 cell surface markers
9. Allogeneic MSCs when transplanted will induce a GvHD
a. If individual has competent immune system, it will kill the MSCs
b. If individual is immunocompromised, MSCs may kill the individual
10. Since tripotent progenitor mesenchymal stem cells did not live up to the promise of articular cartilage repair, Caplan changed the name of the tripotent MSCs to medicinal secretory cells (MSCs) [176-179].
a. These cells were proposed to release paracrine factors to modulate inflammation during the regenerative process
b. The medicinal MSCs express CD73, CD90, CD105, and MHC Class-1 cell surface markers [180-183].
c. Since the cell surface markers are not identical, medicinal MSCs are clearly a different cell type than the tripotent MSC
d. If used autologously, maybe function is correct
e. If used allogeneically, MAY induce GvHD, because of conflicting MHC Class-1 self-recognition molecules
Why MSCs became so popular
Why do you think MSCs became such a dominant topic in regenerative medicine?
Was it because they were easier to obtain, easier to study, safer, or more commercially practical?
1. Political
Initially, I believe it gave politicians something to compare to ESCs.
Table 2. Comparison of ESCs to MSCs
| Attributes | ESCs | MSCs |
| Plasticity | Any somatic cell | Fat, Cartilage, Bone |
| Telomerase enzyme | Present | Absent |
| Proliferation Potential | Unlimited | 70 Population Doublings |
| Age of Individual | Embryo | Adult |
| Location | Inner Cell Mass of Developing Embryo | Adipose Tissue, Bone Marrow, Wisdom Teeth, Umbilical Cord,Umbilical Cord Blood, any organ/tissue with a connective tissue compartment and associated with white fat, hyaline cartilage, and intramembranous bone |
| Self-Recognition Molecules | Allogeneic | Autologous,Allogeneic |
| Graft Vs Host Disease | Yes | No – AutologousYes – Allogeneic |
| Default State | Spontaneous Differentiation | Quiescent |
| Controlled by External Entities | No | Yes |
| Teratoma FormationIn Naïve State | Yes | No |
| Pre-Differentiate | Yes, otherwise Teratoma Formation | No |
| Easier to Study | No | Yes |
| Easier to Process Cells | No | Yes |
| Propagating Ex Vivo for transplants | Yes, but need LIF to prevent premature differentiation | Yes |
11. The Holy Grail ProblemWhy has the field struggled to find one perfect stem cell category?
Except for one group of cells, which we will get to in a few moments, no isolated cells to date filled all parameters for “Holy Grail” Wish List.
Table 3. Holy Grail Wish List
| Parameters | ESCs/iPSCs | MSCs |
| Telomerase Positive | Yes | No |
| Unlimited proliferation Potential | Yes | No |
| Present throughout lifespan of individual | NA | Decrease with increasing age |
| Absent Self-Recognition Molecules | No | No |
| Invisible to Immune System | No | No |
| Will form any somatic cell type | Yes | No,fat, cartilage, bone |
| Does NOT spontaneously differentiate | No | Yes |
| Pre-differentiation is NOT needed | No | Yes |
| Will NOT form teratomas | No | Yes |
| Function controlled by biological agents | No | Yes |
| Homing receptors for damaged cells | ?? | Yes |
| Naïve state forms what is lost or damaged | ?? | Yes |
| Does NOT overgrow existing cells/tissues | ?? | Yes |
| Exosome Production | Yes | Yes |
| Currently, can be propagated to large numbers without mutations | No | No |
| Universal stem cell transplant | No | No |
| Days of shelf-life at 4oC | ?? | ?? |
| Cryopreserved | -196 oC | -196 oC |
| Recovery Viability | ?? | 95% |
| Can be Freeze Dried | No | No |
| Restoration Viability | No | No |
| Can withstand-196 oC to +200 oC | NYD | NYD |
| Bio-printed into 3D Constructs for transplant | NYD | NYD |
Part 3: Introducing aTPSCs as a Fourth Category
1. What is an adult telomerase positive stem cell?
The aTPSCs are actually a category of cells (8 total, divided into 5 subcategories) that are found within all the connective tissues of the body after birth. They are unique in that they retain the telomerase enzyme after birth of the individual. This is unlike differentiated cells and progenitor cells that lose the telomeres enzyme at birth [184]. The aTPSCs are preprogrammed to heal/replace damaged tissues. Their default state in the body is as a dormant quiescent hibernating cell within the connective tissues, where they are maintained throughout the lifespan of the individual. The germ layer lineage stem cells: EctoSCs, MesoSCs, and EndoSCs, prefer an aerobic environment (21% Oxygen saturation) and are located very close to capillaries. TSCs and PSCs prefer an anaerobic environment (5% Oxygen saturation) and are located at maximum distance from the blood vessels.
The aTPSCs only become activated when they receive a signal (chemokine) released from damaged tissues. At which point they begin dividing symmetrically, forming daughter cells. The daughter cells undergo reverse diapedesis into the bloodstream. From there they home to the damaged tissues following an increasing concentration gradient of chemokines. Once on site, local metalloproteinases remove the blocking molecules from their receptor sites and they respond to locally released cues (exosomes/secretomes/biological agents) to restore whatever tissue is lost (Table 4).
Table 4. Attributes of Endogenous Adult Telomerase Positive Stem Cells
|
Attributes
|
TSCs1 |
HLSCs2 |
CLSCs3 |
PSCs4 |
GLSCs5 |
EctoSCs6 |
MesoSCs7 |
EndoSCs8 |
|
Size, microns |
0.1-2.0 |
>2-4 |
>4-<6 |
6-8 |
>8-<10 |
10-12 |
10-12 |
10-12 |
|
0.4% Trypan blue |
Entire Cell Positive9 |
Halo10 Positive, Negative |
Corona11, Positive Negative |
Entire Cell Negative12 |
Entire Cell Negative |
Entire Cell Negative |
Entire Cell Negative |
Entire Cell Negative |
|
Cell Surf Markers Animals |
CEA-CAM-113 |
CEA-CAM-1high SSEA-4low |
CEA-CAM-1low SSEA-4high |
SSEA-414 |
SSEA-4, Thy-115 |
Thy-1 |
Thy-1 |
Thy-1 |
|
Cell Surf Markers Human |
CD66e16 |
CD66ehigh CD10low |
CD66elow CD10high |
CD1017 |
CD10 CD9018 |
CD56, CD90 MHC-119 |
CD13, CD90 MHC-1 |
??? CD90 MHC-1 |
|
Culture Conditions |
Suspen-sion20 Substrate Adhesion 21 |
Substrate Adhesion |
Substrate Adhesion |
Substrate Adhesion |
Substrate Adhesion |
Substrate Adhesion |
Substrate Adhesion |
Substrate Adhesion |
|
Differentia-tion Capabilities22 |
All Cells23 |
Somatic Cells only24 |
Somatic Cells only |
Somatic Cells only |
Somatic Cells only |
Ectoderm Lineage Only25 |
Mesoderm Lineage Only26 |
Endoderm Lineage Only27 |
|
Maximum Proliferation To Date |
>300 Rat Population Doublings |
>300 Rat Population Doublings |
>300 Rat Population Doublings |
>400 Rat Population Doublings |
>400 Rat Population Doublings |
>400 Rat Population Doublings |
>690Human Population Doublings |
>400 Rat Population Doublings |
Table 4. TSCs1, totipotent stem cells; HLSCs2, halo-like stem cells; CLSCs3, corona-like stem cells; PSCs4, GLSCs5, germ layer lineage stem cells; EctoSCs6, ectodermal stem cells; MesoSCs7, mesodermal stem cells; EndoSCs8, endodermal stem cells; entire cell demonstrating Positive9 staining for 0.4% Trypan blue; Halo10, complete peripheral rim of positive trypan blue staining with central area of cell negative for Trypan blue staining; Corona11, crown of positive Trypan blue staining, remainder of cell is negative for Trypan blue staining; entire cell demonstrating Negative13 staining for 0.4% Trypan Blue; CEA-CAM-113, carcinoembryonic antigen-cell adhesion molecule-1; SSEA-414, stage-specific embryonic antigen-4; Thy-115, N-glycosylated glycophosphatidylinositol (=CD90); CD66e16, carcinoembryonic antigen; CD1017, common acute lymphoblastic leukemia antigen (CALLA); CD9018, N-glycosylated glycophosphatidyl-inositol (=Thy-1); MHC-119, self-recognition molecule Major Histocompatibility Complex-Class-1; Suspension20, cells grow in suspension culture only; Substrate Adhesion21, only grows attached to a substrate; Differen Cap22, differentiation capabilities; All Cells32, will form all somatic cells of the body, the gametes (spermatogonia and oogonia), and the nucleus pulposus of the intervertebral disc (the only tissue derived from the notochord in adults); Somatic Cells Only24, will only form somatic cells of the body, will not form gametes, will not form nucleus pulposus of the intervertebral disc; Ecto Lineage Only25, will only form cells of the ectodermal germ layer lineage, will NOT form cells of either the mesodermal or the endodermal germ layer linages; Meso Lineage Only26, will only form cells of the mesodermal germ layer lineage, will NOT form cells of either the ectodermal or the endodermal germ cell lineages; Endo Lineage Only27, will only form cells of the endodermal germ layer lineage, will NOT form cells of either to ectodermal or mesodermal germ layer lineages; NYD28, not yet determined. Reprinted with permission from Young HE, Speight MO. Characterization of endogenous telomerase-positive stem cells for regenerative medicine, a review. Stem Cell Regen Med. 2020; 4(2):1-14 [174].
The name has three important parts, explain what each part means.
Adult: the aTPSCs are found in adult (post-natal, after birth) animals. They have been identified in 15 species of animals, including humans. To be more precise, they have been discovered amphibians (four species of Ambystoma maculatum, annulatum, tigranum, texanum), reptiles (Komodo Dragon), avians (chickens and Wadel Crane), mice, rats, rabbits, cats, dog, sheep, goats, pigs, cows, spectacled bears, horses, and humans. This suggests they are a conserved cell population throughout phylogeny.
Telomerase positive: aTPSCs retain an enzyme called telomerase. Every cell at birth has 70 telomeres at the ends of each of their chromosomes. Telomeres protect the chromosomes from damage. With each cell division one telomere is lost from each chromosome (Hayflick’s Limit). With the telomerase enzyme, after each cell division, a telomere is added back to the ends of the chromosomes. This gives the cells an unlimited proliferation potential. I like to say unlimited, but I only took out a human MesoSC to 690 population doublings (20-year experiment) before the experiment was terminated. At least every 100 population doublings (Z100, Z200, Z300, Z400, Z500, Z600, and Z690) the cells were tested for karyotypic analysis, response to our library of biological agents (proliferative, inductive, progressive, and anti-differentiative) as assessed by the ELICA, identified cell surface markers, karyotyped the cells to determine mutation formation, other characterization parameters and compared to our Z1 MesoSCs from the same batch of cells. The original human MesoSCs were isolated, allowed to proliferate, labeled as Z1, aliquoted at 10^6 cells per ml and cryopreserved using 7.5% ultra-pure DMSO and slow frozen (one degree per minute) and stored at -70oC. A Z1 aliquot was thawed at 37oC, the DMSO removed, plated onto type-1 collagen substrate coated plates, and tested side-by-side with the Z### cells. We could detect no differences between Z1 and any of the Z### with respect to all the parameters examined [106].
Stem Cell: There are actually three general categories of cells in the body [184].
First, are the functional/differentiated cells composed of parenchyma (active functional part of an organ) and the stroma (the connective framework of an organ). They are telomerase negative, and conform to Hayflick’s limit of 70 population doublings before they senesce and die. They compose about 40% of the 37+ trillion cells in the body. Examples of functional cells are pancreatic islet cells, lung pneumocytes, dopaminergic neurons, thyroid follicle cells, rods and cones in the eye, and adipocytes. Usually, any cell with a “cyte” suffix is considered a functional cell.
Second, are the maintenance/progenitor cells. Their function is to maintain the activities of the differentiated cells. When the differentiated cells wear out, senesce, and die, they are replaced with the progenitor cells. The progenitor cells are telomerase negative, and conform to Hayflick’s limit of 70 population doublings before they senesce and die. They compose about 49% of the 37+ trillion cells in the body. Examples of progenitor cells are the multipotent hematopoietic stem cells, tripotent mesenchymal stem cells, bipotent adipofibroblasts, and unipotent adipoblast. Usually, any cell with a “blast” suffix is considered a maintenance/progenitor cell.
Third, are the healing cells/true stem cells. There are eight categories of true stem cells: TSCs, HLSCs, CLSCs, PSCs, GLSCs, EctoSCs, MesoSCs, and EndoSCs, divided into five subcategories: totipotent (TSCs), pluripotent (HLSCs, CLSCs, PSCs, and GLSCs), and germ layer lineage specific (EctoSCs, MesoSCs, and EndoSCs) (Table 4). Their sole function is to heal damaged tissues, restoring the histoarchitecture of the tissue, thereby restoring function. The true stem cells are telomerase positive and therefore have an unlimited proliferation potential. They compose about 1% of the 37+ trillion cells of the body, and can be further divided as 0.3% for each germ layer lineage stem cell, 0.09% for the pluripotent stem cells, and 0.01% for the totipotent stem cells.
Why is it important that these cells are found in the adult body?
Actually, the aTPSCs first appear during embryogenesis.
At the four-cell stage the developing morula divides asymmetrically to form two different populations of cells, based on function. The larger blastomeres are pre-programmed for spontaneous development of the embryo/fetus within the uterus that will become an individual. The blastomeres form all tissues of the conceptus (embryo and extraembryonic membranes), e.g., the notochord, gametes, all somatic cells, the placenta, and their attachment to the embryo/fetus, the umbilical cord. The very small TSCs are pre-programmed for healing, and are tightly regulated by external factors.
Both 4-cell stage blastomeres and TSCs are totipotent, forming all somatic cells of the body, the notochord, gender-specific gametes, and the extra-embryonic membranes (placenta and umbilical cord). Just before derivation of the 16-cell stage morula, the 15th blastomere differentiates into the notochord (the primary inducer of the embryo, secreting sonic hedgehog glycoproteins), and the 16th blastomere which differentiates into gender-specific gametes.
A blastula forms, composed of the trophoblast, e.g., extra-embryonic membranes (future placenta and umbilical cord) and inner cell mass. The pluripotent stem cells (PSCs) appear within the inner cell mass, from which ESCs and iPSCs were derived. The function of the PSCs is to form all somatic cells of the body, under tightly controlled conditions. The function of the ESCs and iPSCS is the same, formation of all somatic cells of the body, but by spontaneous differentiation.
The germ layer lineage stem cells (EctoSCs, MesoSCs, and EndoSCs) appear within their respective germ layer linages: EctoSCs in ectoderm, MesoSCs in mesoderm, and EndoSCs in endoderm. Their function is to form ONLY those cell types within their respect germ layer lineages. Meaning EctoSCs will only form ectodermal-derived cells and will not form mesodermal cells or endodermal cells; MesoSCs will only form mesodermal-derived cells and will not form ectodermal cells or endodermal cells; and EndoSCs will only form endodermal-derived cells and will not form ectodermal cells or mesodermal cells. In other words, trans-differentiation between germ layer lineages does NOT occur naturally.
At birth, the cells that are or will become the functional/differentiated cells and the maintenance/progenitor cells lose the telomerase enzyme and assume Hayflick’s Limit of 70 population doublings before they senesce and die.
At birth, there are cells that have been preprogrammed to heal damaged tissues, the aTPSCs. While few in number, they retain the telomerase enzyme to have unlimited proliferation potential as long as they remain undifferentiated. Once they start the differentiation process, they lose the telomerase enzyme and conform to Hayflick’s Limit, until they senesce and die.
In short, each one of us are carrying our own first aid kit which is present throughout your entire lifespan. Whenever the cells are needed, they are activated, proliferate, home to the damaged cells/tissue and heal, either by repair or regeneration, restoring the original histoarchitecture of the damaged tissues and subsequent function of the organ/tissue.
How does that make them different from embryonic stem cells?
Embryonic stem cells (derived from the inner cell mass, therefore pluripotent) are pre-programmed to spontaneously form all somatic cells of the body. They will do this either in the uterus (normal embryogenesis) or outside the uterus (teratoma formation).
aTPSCs are preprogrammed to HEAL damaged cells and tissues. The healing process is very tightly controlled by an orchestrated series of biological agents. Activation by chemokines, proliferation with symmetrical divisions stimulated by PDGFs, anti-differentiation by ADF bound to receptor sites to prevent premature differentiation, reverse diapedesis of daughter cells into bloodstream, homing to damaged tissue site following concentration gradients of chemokines, metalloproteinase removal of ADF blocking cell surface receptors, continued proliferation stimulated by PDGFs, local exosomes/secretomes attaching to receptors to dictate blueprint for cellular repair of multiple cell types, induction to differentiate into specific cell types, and progression factors to accelerate phenotypic expression of those cell types: thereby forming functional cells for the particular tissue; blood vessels, nerve fibers (Schwann cells forming myelin sheaths, sensory nerve endings, motor end plates), and lymphatic vessels all contained with the connective tissue stroma; and cell specific multipotent, tripotent, bipotent, and unipotent maintenance progenitor cells to maintain the organ.
What is telomerase?
Telomerase is an enzyme that adds one telomere to each chromosome after losing a telomere during cell division. Basically, it re-sets the biological clock of 70 population doublings after each cell division cycle [185].
Why does telomerase matter when we talk about cell division, aging, and repair?
All somatic cells of the body have a biological clock that starts at 70 population doublings with the birth of a human individual. As one ages: i.e., grows in length and width, the somatic cells divide, losing one telomere at each cell division. Adverse life style choices can accelerate cell division in some organs leading to a biological clock of the organ that is less than the chronological age of the individual. Examples would be excessive alcohol consumption leading to liver damage; tachycardia leading to heart failure; and smoking leading to COPD.
Because the aTPSCs exist in a dormant, hibernating, quiescent state, they are not compromised by adverse lifestyle choices, as are telomerase negative somatic cells. Only when they are activated and begin to divide, can the cells be affected by adverse lifestyle choices. In our Informed Consent Guidelines (which we will cover in later podcasts) we have a list of specific dos and don’ts that effect whether a treatment will be a success or a failure [159]. When someone self-treats with aTPSCs (using CNSP) [186] or treated exogenously with fresh isolates of aTPSCs [187], they are adding newborn cells (with a starting Hayflick’s Limit of 70 population doublings) to an older individual. These newborn cells recapitulate the developmental stages of newborn, adolescent, puberal, maturation, sexually mature, etc., as their cell doubling numbers increase. This is seen as a recapitulation of the ECM, PEMs, and functional activities with respect to the different age groups.
For example, if a 70-year-old with white hair was treated with enough aTPSCs, their white hair would revert back to their original hair color. We have seen this phenomenon with multiple individuals after treatment with aTPSCs. If autologous aTPSCs were used, their hair color would revert back to their own original hair color, if allogeneic aTPSCs were used, their hair color would revert back to the original hair color of the donor [186,187].
What makes these cells true stem cells?
The aTPSCs are uniquely different from ESCs, iPSCs, MSCs, iPSCs, MUSE, VSELs, MIAMIs, MAPCs [175], in so many ways: from locations, to existence, preferential environmental conditions, isolation, plating, media composition, propagation without mutations, cell surface markers, response to biological agents (proliferation, progression, induction, anti-differentiation), differentiation potential, sizes, Trypan blue staining, telomerase positivity, unlimited proliferation potential, will form any somatic cell type in the body (plus extra cell types for TSCs), ease of directed transplantations, safety, and efficacy.
What ability they have that ordinary adult cells cannot do? They check “YES” on all the Holy Grail wish list, except the last two, which have not as yet been studied, as well as additional points (Table 5).
Table 5. Holy Grail Wish List (completed)
|
Attributes |
TSCs |
ESCs |
MSCs |
|
Telomerase Positive |
YES |
Yes |
No |
|
Unlimited proliferation Potential |
YES
|
Yes |
No |
|
Present throughout lifespan of individual |
YES |
NA |
Decrease with increasing age |
|
Absent Self-Recognition Molecules |
YES |
No |
No |
|
Invisible to Immune System |
YES |
No |
No |
|
Will form any somatic cell type |
YES |
Yes |
No, fat, cartilage, bone |
|
Does NOT spontaneously differentiate |
YES |
No |
Yes |
|
Pre-differentiation is NOT needed |
YES |
No |
Yes |
|
Will NOT form teratomas |
YES |
No |
Yes |
|
Function controlled by biological agents |
YES |
No |
Yes |
|
Homing receptors for damaged cells |
YES |
?? |
Yes |
|
Naïve state forms what is lost or damaged |
YES |
?? |
Yes |
|
Does NOT overgrow existing cells/tissues |
YES |
?? |
Yes |
|
Exosome Production |
YES |
Yes |
Yes |
|
Currently, can be propagated to large numbers without mutations |
YES |
No |
No |
|
Universal stem cell transplant |
YES |
No |
No |
|
Days of shelf-life at 4oC |
40 |
?? |
?? |
|
Cryopreserved |
-80oC |
-196 oC |
-196 oC |
|
Recovery Viability |
99% |
?? |
95% |
|
Can be Freeze Dried |
NYD |
No |
No |
|
Restoration Viability |
NYD |
No |
No |
|
Can withstand -196 oC to +200 oC |
NYD |
NYD |
NYD |
|
Bio-printed into 3D Constructs for transplant |
NYD |
NYD |
NYD
|
|
Additional Aspects |
|
|
|
|
Trypan Blue Staining |
YES |
No |
No |
|
Size, microns |
0.1-2 |
15-30 |
50-100 |
|
Traverse Blood-Brain Barrier |
YES |
No |
No |
|
Enter Thebesian System of Heart |
YES |
No |
No |
|
Heal heart from inside outward |
YES |
No |
No |
|
Replace all cells in damaged brains |
YES |
No |
?? |
|
Allogeneic cells can add new immune system without bone marrow ablation |
YES |
No |
?? |
|
Restore eyesight in dry macular degeneration |
YES |
No |
?? |
|
Reduce pain and increase ambulation in osteoarthritis |
YES |
?? |
?? |
Why do you think that aTPSCs should be viewed as a fourth category of stem cells?
They are different from any stem cells currently in use, be they ESCs, iPSCs, or Progenitor stem cells (e.g., MSCs, MUSE, VSELs, MIAMIs, MAPCs) [175]. See also Table 5.
What separates the aTPSCs from the other categories?
See Table 5 above, of Holy Grail Wish List + Additional Aspects
Is it their origin, telomerase activity, their potency, their location in the body, their safety profile, or all of the above?
All of the above and more, see Table 5 with respect to biological functional activities.
Function of Biological Agents on aTPSCs
Genomically-labeled clonal populations of adult telomerase positive stem cells, e.g., TSCs, PSCs, EctoSCs, MesoSCs, EndoSCs, and an unlabeled clone of telomerase negative progenitor cells, MSCs, were incubated with various biological agents to determine their function [Table 6]. Four categories of biological agents were recognized: anti-differentiating agents, proliferative agents, progression agents, and inducing agents. Ninety-six well plates were utilized to devise a high throughput screening assay for the testing system. One thousand cells were plated per well on a 1% type-1 collagen substratum for the aTPSCs and on uncoated bare plastic for the tripotent progenitor MSCs. The cells were washed with incomplete culture medium and then incubated with particular agents [175] at physiological concentration, nanogram to microgram quantities per ml, in complete medium containing 10% heat inactivated serum. Medium was changed, dependent on the color of the medium [188]. We utilized 108 immunocytochemical and histochemical staining procedures for specific phenotypic expression markers [Table 3], using an enzyme-linked immuno-culture assay (ELICA) [173], and/or radioimmunoassay (RIAs) [189], to screen aTPSCs and MSC with biological agents to determine their functionality.
Table 5.1. Biological Agents Incubated with TSCs, PSCs, EctoSCs, MesoSCs, EndoSCs, and MSCs to Determine Function Activities.
|
Agent |
Name |
Activity |
|
|
|
Anti-Differentiation |
|
LIF |
Leukemia Inhibitory Factor |
Inhibits differentiation of TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs. Has slight inhibitory effect on MSCs |
|
ADF |
Anti-Differentiation Factor |
Inhibits differentiation of TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs. Has slight inhibitory effect on MSCs |
|
CAF |
Caffeine |
Inhibits differentiation of TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs. Has NO inhibitory effect on MSCs |
|
SIF |
Scar Inhibitory factor |
Inhibits differentiation of TSCs, PSCs, and MesoSCs from scar tissue formation (abnormal fibroblast/fibrocyte and abnormal ECM), both in vitro and in vivo. No effect on EctoSCs, EndoSCs, or MSCs. |
|
|
|
Proliferation |
|
PDGF-AA |
Platelet-Derived Growth Factor-AA |
Induces proliferation in TSCs, PSCs, EctoSCs, MesoSCs, EndoSCs, and MSCs |
|
PDGF-AB |
Platelet-Derived Growth Factor-AB |
Induces proliferation in TSCs, PSCs, EctoSCs, MesoSCs, EndoSCs, and MSCs |
|
PDGF-BB |
Platelet-Derived Growth Factor-BB |
Induces proliferation in TSCs, PSCs, EctoSCs, MesoSCs, EndoSCs, and MSCs |
|
|
|
Progression |
|
IGF-1 |
Insulin-Like Growth Factor-1 |
Acts as a progression agent to accelerate differentiation with subsequent expression of phenotypic markers in MSCs, indicative of fat, cartilage, and bone. Has NO effect on TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs. |
|
IGF-2 |
Insulin-Like Growth Factor-2 |
Acts as a progression agent to accelerate differentiation with subsequent expression of phenotypic markers in MSCs, indicative of fat, cartilage, and bone. Has NO effect on TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs. |
|
INS |
Insulin |
Acts as a progression agent to accelerate differentiation with subsequent expression of phenotypic markers in MSCs, indicative of fat, cartilage, and bone. Has NO effect on TSCs, PSCs, EctoSCs, MesoSCs, and EndoSCs. |
|
|
|
Induction |
|
BMP-2 |
Bone morphogenetic protein-2 |
Induces endochondral ossification in TSCs, PSCs, MesoSCs, MSCs; no inductive effect on EctoSCs or EndoSCs. |
|
OMP/CM |
Osteogenic morphogenetic protein/Conditioned Medium |
Induces membranous ossification in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. |
|
CMP/CM |
Cartilage morphogenetic protein/ Conditioned Medium |
Induces cartilage formation in TSCs, PSCs, MesoSCs, and MSCs (hyaline cartilage); no inductive effect on EctoSCs or EndoSCs. |
|
AdipMP/CM |
Adipocyte morphogenetic protein/ Conditioned Medium |
Induces formation of white adipose tissue (fat) formation in TSCs, PSCs, MesoSCs, MSCs; no inductive effect on EctoSCs or EndoSCs. |
|
BMP-4 |
Bone morphogenetic protein-4 |
Induces endothelial cell formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
a-FGF |
Acidic fibroblast growth factor |
Induces endothelial cell formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
ECGF |
Endothelial cell growth factor |
Induces endothelial cell formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
VEGF |
Vascular endothelial growth factor |
Induces endothelial cell formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
BVMP/CM |
Blood Vessel Morphogenetic protein/ Conditioned Medium |
Induces vasculogenesis in TSCs, PSCs, and MesoSC; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs. |
|
FMP/CM |
Fibroblast morphogenetic protein/ Conditioned Medium |
Induces normal fibroblast/fibrocyte formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs. |
|
ScFMP |
Scar Fibroblast morphogenetic protein |
Induces abnormal fibroblast/fibrocyte formation with aberrant extracellular matrix, equivalent in all respects to scar tissue formation in vivo in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. |
|
TGF-b |
Transforming growth factor beta |
Induces scar tissue formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
b-FGF |
Basic fibroblast growth factor |
Induces scar tissue formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
EPO |
Erythropoietin |
In conjunction with c-Kit & IL-6, induces formation of RBCs in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
c-Kit |
c-Kit |
In conjunction with EPO & IL-6, induces formation of RBCs in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
IL-6 |
Interleukin-6 |
In conjunction with EPO & c-Kit, induces formation of RBCs in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. Slight proliferative effect on MSCs |
|
NGF |
Nerve growth factor |
Induces formation of neuronal lineage cells (neurons, oligodendrocytes, astrocytes, Schwann cells, ganglion cells) in TSCs, PSCs, EctoSCs; no inductive effect on MesoSCs, EndoSCs, or MSCs |
|
BrnMP/CM |
Brain Morphogenetic Protein/ Conditioned Medium |
Induces formation of neurons, astrocytes, and oligodendrocytes in TSCs, PSCs, and EctoSCs; no inductive effect on MesoSCs, EndoSCs, or MSCs. |
|
HGF |
Hepatocyte growth factor |
Induces formation of liver lineage cells (oval cells, canalicular cells, biliary cells) in TSCs, PSCs, EndoSCs; no inductive effect on MesoSCs, EctoSCs, or MSCs |
|
LivMP/CM |
Liver Morphogenetic Protein/ Conditioned Medium |
Induces formation of multiple liver cells (oval cells, canalicular cells, biliary cells) in TSCs, PSCs, and EndoSCs; no inductive effect on EctoSCs, MesoSCs, or MSCs |
|
LngMP/CM |
Lung Morphogenetic Protein/ Conditioned Medium |
Induces formation of type-1 alveolar cells in TSCs, PSCs, and EndoSCs; no inductive effect on EctoSCs, MesoSCs, or MSCs |
|
PanMP/CM |
Pancreatic Morphogenetic Protein/ Conditioned Medium |
Induces formation of pancreatic ductal cells, alpha-cells, beta-cells, and delta-cells in TSCs, PSCs, and EndoSCs; no inductive effect on EctoSCs, MesoSCs, or MSCs. |
|
KerMP/CM |
Keratinocyte Morphogenetic Protein/ Conditioned Medium |
Induces keratinocytes in TSCs, PSCs, and EctoSCs; no inductive effect on MesoSCs, EndoSCs, or MSCs. |
|
SpCM |
Spermatogonia Conditioned Medium |
Induces spermatogonia in TSCs; no inductive effect on PSCs, MesoSCs, or MSCs. |
|
SkMMP/CM |
Skeletal Muscle Morphogenetic Protein/ Conditioned Medium |
Induces skeletal muscle cells in TSCs, PSCs, and MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. |
|
SmMP/CM |
Smooth muscle morphogenetic protein/ Conditioned Medium |
Induces smooth muscle cells in TSCs, PSCs, and MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. |
|
CdMMP/CM |
Cardiac muscle morphogenetic protein/ Conditioned Medium |
Induces cardiac muscle cells in TSCs, PSCs, and MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs. |
|
TenMP/CM |
Tendon morphogenetic protein/ Conditioned Medium |
Induces tendon formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs |
|
LigMP/CM |
Ligament morphogenetic protein/ Conditioned Medium |
Induces ligament formation in TSCs, PSCs, MesoSCs; no inductive effect on EctoSCs, EndoSCs, or MSCs |
Table 5.1. Reprinted with permission Young, et al. Adult reserve stem cells and their potential for tissue engineering. Cell Biochem Biophys. 2004; 40(1):1-80 [106]; Young HE. Carcinoembryonic antigen-cell adhesion molecule-1 and stage-specific embryonic antigen-4 are present in the reproductive organs of adult mammals. GSC Advanced Research and Reviews. 2025; 23(03): 149-157 [130].
Part 4. Telomerase, aging, and Hayflick’s Limit
For a general audience can you explain what the Hayflick’s Limit?
Hayflick (1965) published that the maximum number of population doublings that would occur in adult human cells was 70 (from birth). This became known as Hayflick’s Limit [190].
Why do most adult cells only divide certain number of times?
Adult cells, e.g., functional cells and maintenance cells were preprogrammed during embryogenesis to lose the telomerase enzyme at birth and assume a lifespan of 70 population doublings before senescence and cell death. This has come to be known as Hayflick’s Limit.
Is it fair to say that most cells in the body have a biological clock?
Yes, every cell in the body (e.g., healing cells, progenitor cells, and differentiative cells) has a biological clock of 70 population doublings.
What sets aTPSCs apart from differentiated cells and progenitor cells is the presence of the telomerase enzyme that re-sets their biological clock to 70 population doublings after each cell division.
If so, what happens when the clock runs out?
For somatic cells, when the clock runs down to zero, the cell senesces and dies.
What is the difference between a telomerase positive cell and a telomerase negative cell (with respect to function)?
An aTPSC has the ability to form multiple cell types. The extreme case are the TSCs, which will form all somatic cells of the body, gender-specific gametes, nucleus pulposis of intervertebral disc, placenta, and umbilical cord. Intermediate case are the PSCs which will form all somatic cells of the body, and then the germ layer lineage stem cells that will only form cells types within their respective germ layer lineages (Figure. 4).
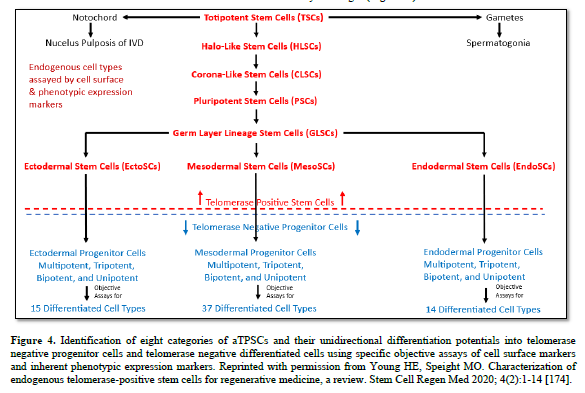
Telomerase negative differentiated cells and progenitor cells will ONLY form several to one specific cell type.
Why does the difference matter in regenerative medicine?
In regenerative medicine one wants to form multiple cell types to completely restore the histoarchitecture of the tissue/organ and thereby restore its function.
If you use a differentiated cell (ex, cardiomyocyte) induced from an iPSC, to repair a myocardial infarction, it may function short term. But, with no connective tissue cardiac skeleton to attach to, you only have fibrillation of the heart muscle, there is no coordinated contraction of pumping blood from the heart. Likewise, if the vasculature is not restored the newly transplanted cell will die because of absence of nutrients and removal of waste products.
In a second example, if you use a progenitor cell (tripotent MSC, that only forms white fat, hyaline cartilage, and intramembranous none) and place it into an articular joint, if you are lucky only hyaline cartilage will form. But hyaline cartilage is not designed for weight bearing, sheer force, tension, and other functions of articular cartilage, and it will fail.
If you want to regenerate all cell types to restore an organ, one needs a cell or cells that has/have that capability, aTPSCs.
The videos describe aTPSCs as being able to divide without the same limits as ordinary adult cells.
How should people understand the idea without misunderstanding it?
Every somatic cell in the body has a biological clock of 70 population doublings. Once their biological clock reaches zero, the cell senesces and dies
What sets aTPSCs apart from differentiated cells and progenitor cells is that their biological clock is re-set to 70 population doublings after each cell division. This gives them to have an unlimited proliferation potential as long as the cells stay undifferentiated. Once these cells commit to a specific cell lineage, they lose the telomerase enzyme, and assume all the characteristics of progenitor cells, including having a biological clock of 70 population doublings.
When people hear that a cell can continue to divide, they may immediately think of cancer cells.
How do you explain the difference between normal telomerase positive cells and a cancer cell.
aTPSCs are tightly controlled using specific proliferation agents (PDGFs). If you want the aTPSCs to proliferate, add PDGF. If you want them to stop proliferating, remove the PDGF. It is that simple.
Cancer cells have gate keeper genes (P53 and P16, among others) that control the ability of the cell to stop division during the cell cycle. If the gate keeper genes are mutated so they do not function, there is uncontrolled cellular proliferation. Once it is turned on, it usually cannot be turned off [191].
Is the key difference that aTPSCs are controlled by the body’s normal biological signals?
There are four biological activities that we have detected: proliferation controlled by PDGFs; progression controlled by IGF-1, IGF-2 and insulin; induction controlled by multiple cell-specific inductive agents (recombinant proteins, morphogenetic proteins, and cell-specific exosomes/secretomes); and anti-differentiation, controlled by either LIF (cell number specific), ADF (inductive factor concentration specific), Caffeine (>95-mg/day), or SIF (scar inhibitory factor) (Table 5) [106,130,175].
How should we think about the balance between regenerative potential and biological control?
Biological control allows regenerative potential to occur through a well-tuned orchestrated series of steps leading to complete restoration of the damaged cells/tissues, thus restoring function.
Part 5: Where are aTPSCs Found in the Body
At a high level, where are aTPSCs located in the adult human body?
Maternal aTPSCs are located within specific connective tissue niches throughout the body. Because the germ layer lineage stem cells, EctoSCs, MesoSCs, and EndoSCs, prefer an oxygenated (aerobic) environment, they are located nearest to the capillaries throughout the connective tissues. And, because the TSCs and PSCs are preferential to a deoxygenated environment (anaerobic), they are located further away from the capillary vascular supply.
The video describes the cells being located in the connective tissues
Why is the connective tissue such an important place for a repair system to exist?
Every organ and tissue in the body is composed of parenchyma (active/functional part of the organ) and stroma (the connective tissue framework of the organ). Located in the connective tissue stroma, gives the aTPSCs immediate access to any trauma that occurs with respect to the organ. Initially, organ area-resident TSCs, PSCs, and MesoSCs are involved in the formation of the transitional scar (band-aid to wall off an external hostile environment from the fragile internal environment). This occurs under the direction of TGF-beta and basic-FGF, released from platelets. Later, when these two biological agents dissipate, additional aTPSCs, migrating through the ECM and arriving via the vasculature, are involved in the later stages of tissue repair. Regenerating/restoring the damaged area to its original histoarchitecture.
Is it fair to think that aTPSCs are a part of the body’s normal repair network?
What would you add to make the analogy more scientific accurate?
Yes, they form the majority of the cells involved in the normal repair (healing) network.
Progenitor/maintenance cells are programmed to replace worn out functional cells.
Differentiated/functional cells are programmed for specific functions for every organ throughout the body.
The videos use the term quiescent, meaning the cells are resting or quiet.
Why would the body keep powerful stem cells in a quiet state most of the time?
Think about it. Why would you need activated aTPSCs if the person was healthy (HF16, HF17)? You wouldn’t. They are activated when there is either a chronic disease (HM1) or a catastrophic injury (HF14, HF15) to the body. And the body decides what exactly that means (Figure. 5).
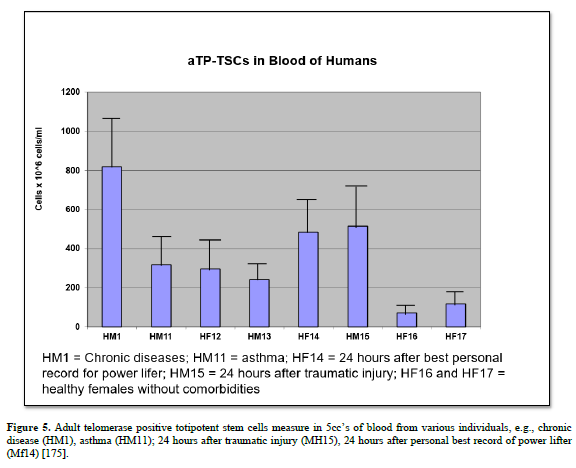
Without getting too deep into the methodology, what kinds of signals normally tell the body that repair is needed?
Are we talking about injury, inflammation, tissue stress, or damage signals?
When a tissue is damaged, creating dead tissue, inflammation occurs. This process recruits cells of the innate immune system (macrophages, NK-cells, neutrophils) to migrate to the damaged tissue site and remove the dead and dying cells from the area. This occurs beneath the transitional scar, eventually forming a sterile area for restoration of the tissues to occur. During the innate immune system phase, substances called chemokines (ADF, migratory factors, and others), are released from the damaged tissues into the surrounding area (ECM and vasculature) in a concentration gradient 360-degree fashion along an X, Y, and Z axis. As the chemokines come in contact with quiescent aTPSCs the activation process occurs: first, there is proliferation: symmetrical division of the resident maternal cell into a migrating daughter cell and a resident maternal cell; second, there is masking of the exosome/secretory receptors with ADF during their journey to the wound site to prevent premature differentiation; third, there is migration of the aTPSC daughter cells through the adjacent ECM and reverse diapedesis of the daughter cells into the bloodstream to arrive at the wound site.
Part 6. Potency and Differentiation
The videos mentioned words like totipotent and pluripotent.
Can you explain those terms in simple language?
Totipotent means a cell has the ability to form all somatic cells of the body, gender-specific gametes (sperm and ova), the nucleus pulposus of the intervertebral disc (the only adult functional derivative of the notochord), and the extra-embryonic membranes (placenta and umbilical cord). Examples: Zygote, 4-cell stage blastomeres, and TP-TSCs (Figure. 4) [174].
Pluripotent means a cell has the ability to form all somatic cells of the body. Examples: cells of the inner cell mass of the developing embryo; epiblast; ESCs, iPSCs, and TP-PSCs (Figure. 4) [174].
How should people think about the hierarchy of stem cells?
Is it like starting with a more flexible cell that becomes more specialized over time?
Actually, the aTPSCs demonstrate a similar hierarchy as the developing zygote that eventually forms the embryo/fetus.
Lineage Map of Embryonic Development (Figure. 2).
The most primitive aTPSC is the TSC (totipotent stem cell) it has the most plasticity at forming all cell types (Figure. 4).
It can differentiate into gametes, NP of IVD, placenta, and PSCs
Then follow through the diagram to more specialized functional cells
What does differentiation mean?
Differentiation means a transition from a more primitive cell to a more specialized cell
How does a cell become more specialized?
There is a series of transitions, from left to right (Figure. 6).
Diagrammatic Summation of Unidirectional Differentiation of aTPSCs
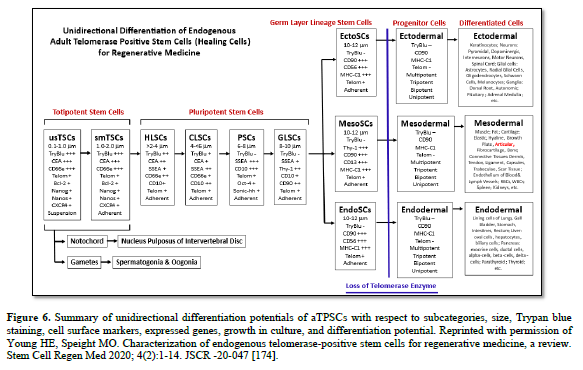
The differentiation of aTPSCs to differentiated cells is a transition through multiple cell types, based on size, Trypan blue staining, cell surface markers, expressed genes, adherent growth in culture, appearance in culture, and differentiation potential (Figure. 6) [174].
The videos described differentiation as a one-way path
Can you explain what that means?
Once a cell becomes specialized, what does that mean?
In the simplest of terms, it means that the specialized cell has lost the capacity to form other specialized cell types, progenitor cells, or stem cells (Figure. 6). In other words, it is unidirectional.
At the general level, when the body repairs a tissue, it is replacing damaged cells, supporting existing cells, changing the local environment, or some combination?
The pre-programmed job of progenitor cells is to replace worn out differentiated cells to maintain function of the organ/tissue.
The job of the stem cells (aTPSCs) is to repair a traumatic insult to the body replacing what was damaged or lost.
Yes, potency matters.
Why does the level of potency matter in regenerative medicine?
Most organs and tissues are a combination cells from ectoderm, mesoderm, and/or endoderm germ layer linages.
The more potent (less differentiated) a stem cell is the better able it is to replace all the damaged tissues, whether they are ectodermal, mesodermal, and/or endodermal in origin.
For example, if one needed to repair a lung the basic cells involved would be pneumocytes (endoderm), blood vessels (mesoderm), and nerves (ectoderm).
A cell that forms only fat, cartilage, and bone (tripotent progenitor MSC) would not form any of these cell types and therefore should not be used.
On the other hand, due to size and differentiation potentials; pneumocytes can be formed from TSCs and PSCs; blood vessels can be formed from TSCs, PSCs, and MesoSCs; and nerves can be formed from TSCs, PSCS, and EctoSCs. This was the methodology we used to regenerate lung tissue for COPD and IPF patients
Why is it important to understand what a stem cell can realistically become?
The originally labeled “adult stem cells” were actually telomerase negative progenitor cells specialized for specific cell types or specific lineages of cells:
Part 7. Why the field has overlooked aTPSCs
A skeptical physician might ask:
If these cells are so important, why haven’t I heard about them?
How would you answer that?
Because of existing dogma, true adult stem cells were thought not to exist (Dogma).
Therefore, to regenerate new cells to replace dead and dying cells, one needed to find a stem cell population that could perform that function.
In 1991, Arnold Caplan published in article in Science concerning the discovery of mesenchymal stem cells (MSCs) from bone marrow in adults. These stem cells would form fat, cartilage, and bone. The MSCs had a defined lifespan of 70 population doublings before it would senesce and die. The adult stem cells decreased with increasing age of the individual.
Other adult progenitor cells were published: hematopoietic, neural, hepatic, etc., with the same basic attributes as MSCs. These were progenitor cells, masquerading as stem cells, were designated as “adult stem cells” to fit that narrative.
In 1998, James Thomson published an article in Science concerning the derivation of human embryonic stem cells from the inner cell mass of the developing embryo. These were stem cells that were pluripotent and could spontaneously form all cells in the body. They also expressed the telomerase enzyme, which allowed them unlimited proliferation.
Since there was a moratorium on studying humans undergoing embryogenesis, Dr. Thomson’s original game plan was to use these cells to study differentiation during embryogenesis, to be able to correct health problems in individuals before birth.
Then someone had the “bright idea” that these same ESCs could repair damage in adults after birth.
That bright idea created a “firestorm” with the Vatican, right politically-leaning individuals, etc., stating that it is morally and ethically WRONG to destroy one potential human being to heal another, embryos have rights too.
Then the debate was on, which one is better ESCs or Adult stem cells.
By order of the sitting POTUS, NIH denied funding for ESC research in any form, so these adult stem cells, with MSCs in the forefront became the NIH fundable stem cells of choice.
Next, other adult stem cells, such as VSELs, MUSE, MIAMIs, and MAPCs, as variations of the MSCs. They are apparently a type of lineage-committed progenitor cell, that are telomerase negative, have a 70-population doubling biological clock, and decrease in number with increasing age of the individual [175].
Do you think the field became more focused on certain categories, like ESCs, iPSCs, and MSCs, and simply did not look closely enough at this fourth category.
Where articles are published
Key point here is where the research was published: Science, Nature, Cell, PNAS, JAMA,
Most scientists, physicians, lay people, at least in USA, only read articles from these journals with the mindset that they are the most important work why waste their time with lesser journals. And these journals published on ESCs, iPSCs, and MSCs.
I publish my research in 2nd, 3rd, and 4th, tier journals.
Were aTPSCs difficult to identify or study because they are rare?
No, I already knew what to look for and where to find them from my work with adult salamanders, that wasn’t the problem
Did the technology or methodology have to catch up before they could be properly characterized?
The problem was that existing technology and methodologies were not available at the time I stated my studies, nor during my studies.
So, I developed what I needed when I needed it.
I screened the Developmental Hybridoma Bank of Monoclonal antibodies for ones that would attach to these cells: CEA-CAM-1 (TSCs), SSEA-4 (PSCS), Thy-1 (EctoSCs, MesoSCs, and EndoSCs)
Unfortunately, not all antibodies worked with the same fixatives or any fixatives, so I developed the ELICA fixative [173]:
I developed the ELICA – a high throughput screening assay that [173]:
Since the aTPSCs did not follow the Guidelines of ATCC “Bible” for the isolation and culturing of cells, I had to develop everything from scratch: Isolation, Buffer specificities, Medium, Plating, Propagation, Release from culture vessels, Activation Ex Vivo, Replating, Cryopreservation, Viability testing [192-198].
I screened Beckton-Dickinson (BD) Cluster of Differentiation markers for human cells. Then performed cell sorting with the antibodies [175,194]:
Do you think part of the problem is that the stem cell terminology has become confusing?
Yes. Best example is the acronym MSCs
Originally MSCs (mesenchymal stem cells) were a tripotent progenitor cell that would form fat, cartilage, and bone. They are CD90, CD105, CD123, CD166, MHC Class-1 positive.
Name change to MSCs (medicinal secretory cells) that modulated the immune system during repair, they are CD73, CD90, CD105, and MHC Class-1 positive. Clearly a different cell from the original tripotent MSC.
Then there are MSCs (marrow stromal cells) that support cells undergoing hematopoiesis. The are CD29, CD90, CD146, CD166, CD271, MHC Class-1. Also, a different cell from the original tripotent MSC
But all use the same acronym MSC
If a clinician is not familiar with the differences in MSCs in the literature, they may isolate the medicinal MSCs using the protocol for the tripotent MSC. And then use the tripotent MSC to modulate the immune response during repair of damaged tissues.
This occurred and presented at a meeting of the FDA by patients having failed treatments.
How should the field become more precise in how it names and classifies cells?
Personal opinion, the stem cells should be named according to their unique characteristics and functional capabilities. For the aTPSCs that would be:
Many doctors are familiar with MSCs
What is the simplest way to explain that aTPSCs are not the same thing as MSCs?
Table 6. Comparison Contrast of aTPSCs vs Tripotent MSC vs Medicinal MSC.
|
Characteristics |
aTPSCs |
Tripotent MSC |
Medicinal MSC |
|
Size, microns |
0.1-12 |
15-30 |
15-30 |
|
Telomerase |
Present |
Absent |
Absent |
|
Proliferation |
Unlimited |
70 Pop. Doub. |
70 Pop. Doub. |
|
Lifespan |
Present Throughout |
Decrease with age |
Decrease with age |
|
Unique CD Markers |
CD66e, CD10 |
CD105, CD123, CD166 |
CD73 |
|
Repair |
All somatic cells |
Fat, cartilage, bone |
Immune Support |
|
Regeneration |
All somatic cells |
Fat, cartilage, bone |
Immune Support |
|
# Cell types formed |
70+ |
3 |
none |
The stem cell field has a lot of hype
How do we introduce a new stem cell category responsibly without over promising?
We publish the findings from human clinical trials and let the data speak for itself.
Table 7. Results from IRB-Approved Clinical Study Protocols for Fresh Isolate aTPSCs.
|
Trials |
Clinical Trial |
Sample Size, n= |
Adverse Events |
Description |
Efficacy |
|
1 |
Osteoarthritis |
6 |
None |
Decreased pain, increased ambulation |
100% |
|
2 |
Systemic Lupus |
1 |
None |
Rescued from death, increased organ functioning from less than 25% to ~80%, 10+ years |
100% |
|
3 |
Idiopathic Pulmonary Fibrosis |
2 |
None |
Increased pulmonary function in one participant from 14% to 27%, and then stabilized at 25% for 8+ years. In other participant from <25% to ~70% for almost 10+ years |
100% |
|
4 |
Chronic Obstructive Pulmonary Disease |
51 |
None |
48 participants demonstrated increase in lung function, one participant for 8+ years. Three participants showed no effect to treatment, but did not follow informed consent guidelines |
94% |
|
5 |
Celiac Disease |
1 |
None |
Completely reversed symptoms of celiac disease, went from 1:73 titer to 1: |
100% |
|
6 |
Cardiovascular Disease |
2 |
None |
One participant had myocardial infarction six years prior to treatment initiation. 1st Treatment raised cardiac output from <25% to 35%, 2nd Treatment from 35% to 45%; Other participant raised cardiac output from <25% to ~70% |
100% |
|
7 |
Cardiovascular Disease with CN-SP only |
1 |
None |
One participant with <10% cardiac output, ingested CN-SP only. Within 6 months, cardiac output rose to 35%. +6 more months, cardiac output >45%. |
100% |
|
8 |
Age-Related Dry Macular Degeneration |
4 |
None |
Two participants completely reversed symptoms. Other two participants Treatments did not work, but they did not follow informed consent guidelines |
50% |
|
9 |
Alzheimer’s Disease |
4 |
None |
Two participants completely reversed symptoms. Other two participants Treatments did not work, but they did not follow informed consent guidelines |
50% |
|
10 |
Parkinson’s Disease |
12 |
None |
10/12 showed reversal of symptoms 1st month after Treatment. At 7 & 14-months post-Tx 2/12 regressed at slower rate than before treatments began; 4/12 remained in stasis; 4/12 normal or near normal. 2/10 – no response, did not follow protocol |
66% |
|
11 |
Traumatic Blindness |
1 |
None |
From completely blind to shades of black and gray (partial restoration of ‘night’ vision) after two Treatments. |
100% |
|
12 |
Traumatic Spinal Cord Injury |
1 |
None |
From complete paraplegia from T12 and below, to regain of bladder/bowel function after two treatments. |
100% |
|
13 |
Chronic Inflammatory Demyelinating Polyneuropathy |
3 |
None |
Inability to walk prior to treatments. 2/3 demonstrated ability to walk unassisted/ 1/3, no change – did not following informed consent guidelines |
66% |
|
14 |
Stroke |
1 |
None |
Decreased cognition pre-treatment. Post-treatments showed increasing gain of cognitive function |
100% |
|
15 |
Traumatic Brain Injury |
1 |
None |
Decreased cognitive function, no ambulation on limbs on one side of body. After two treatments showed increased cognition & ability to move all limbs |
100% |
|
16 |
Multiple Sclerosis |
3 |
None |
1st participant pre-treatment: decreased cognitive function, motorized wheelchair and on ventilator 24/7; Post x 2 treatments – increased cognitive function, walk with leg braces, drove vehicle, breathing own for 4+ years. 2nd & 3rd participants – no effect, did not follow informed consent guidelines |
33% |
|
17 |
Amyotrophic Lateral Sclerosis |
2 |
None |
Two participants – one showed stasis to slow decline for 4+ years; other currently in stasis for 11+ years |
100% |
|
18 |
Chronic Kidney Disease |
1 |
None |
Reversed symptoms of kidney failure and restored kidney function for 3+ years |
100% |
|
|
Totals |
97 |
Safe |
Average Efficacy |
86% |
Table 7. Clinical trial results using fresh isolates of aTPSCs demonstrated that cumulative treatment of 97 people with 100% safety and 86% efficacious at reversing their signs and symptoms. Reprinted with permission from Young HE. Fresh Isolate Adult Telomerase Positive Stem Cells: An addition to Embryonic Stem Cells (ESCs), Induced Pluripotent Stem Cells (iPSCs), and/or Mesenchymal Stem Cells (MSCs) for Regenerative Medicine. GSC Advanced Research and Reviews. 2023; 16(1):066-081 [187].
Combinatorial Nutraceutical Supplement Pill (CNSP) aTPSC Therapy
CNSP was designed to try and mimic the fresh isolate aTPSC therapy, but in situ without the stem cells ever leaving the body to be activated. The fresh isolate aTPSC therapy utilizes a bolus of activated stem cells given at directed sites and by intravenous infusion. The CNSP therapy utilizes a continuous release of activated aTPSCs mobilized into the bloodstream 24/7.
Individuals were requested to ingest CNSP continuously throughout their respective trial. CNSP was designed to 1. induce proliferation of connective tissue resident aTPSCs, 2. mobilize the proliferated aTPSCs into the bloodstream, 3. increase circulation throughout the body, 4. unmask homing receptors for damaged tissues, 5. unmask receptors for local environmental inductive agents (exosomes), 6. support a strong innate immune system, and 7. prevent tissue overgrowth. The dosage of CNSP is based on body weight of the individual. One capsule per fifty pounds body weight equals maintenance dose, whereas one capsule per 25 pounds body weight equals healing dose. Individuals were requested to ramp-up dosage of CNSP to optimal dosage for individual. Week one, one capsule of CNSP per day. Week two, two capsules of CNSP per day. Week three, three capsules of CNSP, so on and so forth to optimal healing dose. And then optimum healing dose thereafter [Table 8].
Table 8. Results from IRB-Approved Clinical Study Protocols for aTPSC Therapy with CNSP.
|
Trials |
Clinical Trial |
Sample Size, n= |
Adverse Events |
Description |
Efficacy |
|
1 |
Degenerative Disc Disease, Back Pain, Scoliosis |
1 |
None |
Individual presented with back pain and scoliosis secondary to degenerative disc disease. Treated with fresh isolates, chiropractic manipulations, and CNSP – complete resolution of symptoms. |
100% |
|
2 |
Lupus-Induced Glaucoma |
1 |
None |
CNSP + surgery to reduce internal eye pressures from 30+ bilateral to normal: right eye 10 and left eye 12, CNSP maintaining eye pressures, 2+ years and counting |
100% |
|
3 |
Back Pain |
1 |
None |
Fresh Isolates + Chiropractic manipulations + CNSP resulted in reduced pain |
100% |
|
4 |
Degenerative Disc Disease |
1 |
None |
Fresh Isolates + Chiropractic manipulations + CNSP resulted in restoration of articular cartilage at facet joints; restoration of topography of intervertebral discs; allow restoration of movement |
100%
|
|
5 |
Induced Scoliosis |
1 |
None |
Fresh isolates + chiropractic manipulations + CNSP resulted in restoration of vertical alignment of vertebral column |
100% |
|
6 |
Systemic Lupus Erythematosus (SLE) |
1 |
None |
Fresh isolates + CNSP maintaining stasis – 24+ months and counting |
100% |
|
7 |
Idiopathic Pulmonary Fibrosis (IPF) |
1 |
None |
Fresh isolates + CNSP maintaining FEV1 at ~70%, with O2 saturation at 99-100%, 24+ months and counting |
100% |
|
8 |
Cardiomyopathy |
1 |
None |
Maintaining cardiac output at ~70% with S/D of 106/50 with HR of 60, 24+ months and counting |
100% |
|
9 |
Vision |
20 |
None |
Increased color acuity, colors are brighter and sharper |
100% |
|
10 |
Brain Fog |
20 |
None |
Decreased brain fog |
100% |
|
11 |
Cognition |
20 |
None |
Increased cognition |
100% |
|
12 |
Energy Level |
20 |
None |
Increased energy levels |
100% |
|
13 |
Fatigue/Tiredness |
20 |
None |
Less Fatigue and tiredness |
100% |
|
14 |
Generalized Aches & Pains |
20 |
None |
Gone, systemic pain free |
100%
|
|
15 |
Depression |
20 |
None |
Decreased depression |
100% |
|
16 |
Outlook on Life |
20 |
None |
Better outlook on life |
100% |
|
17 |
Weight Loss |
2 |
None |
Weight loss, 30 lbs. in female and 15 lbs. in male – 6 months post CNSP ingestion |
100% |
|
18 |
T12 Paraplegic, loss of both motor and sensory functions below waist |
1 |
None |
Regained sensory input from waist to top of knees; can move thighs, both flexion and extension – 6 months after starting CNSP |
100% |
|
19 |
Hip Pain |
1 |
None |
No more hip pain one week after starting CNSP, continues pain free – 24+ months on CNSP |
100% |
|
20 |
Power Lifter |
1 |
None |
Decrease in times between obtaining best personal records – 12+ months on CNSP |
100% |
|
21 |
Post Myocardial Infarction |
1 |
None |
<10% cardiac output after MI, could not walk ten steps without passing out, placed on national heart registry for transplant. Within 6 months on CNSP, cardiac output raised to 35%. +6 more months, cardiac output >45%. Playing 9-holes golf weather permitting. |
100% |
|
22 |
Open heart surgery to replace mitral valve |
1 |
None |
Maintaining stasis after heart surgery – 9+ years |
100% |
|
23 |
Shingles |
1 |
None |
Reduced pain, itching, burning sensations. On a pain scale of 0 to 10; before treatment subjective pain was 10/10; currently subjective pain is 2/10 |
100% |
|
24 |
Squamous Cell Carcinoma |
1 |
None |
100% healing of wound after excision of cancer |
100% |
|
25 |
Injured Shoulder / Rotator Cuff |
1 |
None |
Reduced pain to about 5% and increased function to 90% |
100% |
|
26 |
Rheumatoid Arthritis |
1 |
None |
Reduced pain, increased ambulation |
100% |
|
27 |
Hair color |
2 |
None |
Returning to original adolescent hair color |
100% |
|
28 |
Wrinkles & Crepe Skin |
2 |
None |
Returning to original adolescent facial topography |
100% |
|
|
Total |
43 |
Safe |
Average Efficacy |
100% |
Table 8. Clinical trial results using CNSP for the activation of aTPSCs in situ demonstrated that cumulative treatment of 43 people with 100% safety and 100% efficacious at reversing their signs and symptoms. Reprinted with permission from Young HE. Combinatorial nutraceutical supplement pill (CNSP) stimulates naïve adult telomerase positive stem cells in-situ to reverse signs and symptoms in multiple health conditions. GSC Advan Res Rev. 2024; 20(02): 047-056 [201].
Total cumulative results from fresh isolates & CNSP for 140 participants showed that the treatments were safe 100% of the time, average efficacy is 89%. Reprinted with permission from Young HE. Fresh Isolate Adult Telomerase Positive Stem Cells: An addition to Embryonic Stem Cells (ESCs), Induced Pluripotent Stem Cells (iPSCs), and/or Mesenchymal Stem Cells (MSCs) for Regenerative Medicine. GSC Advanced Research and Reviews. 2023; 16(1):066-081 [187]; Young HE. Combinatorial nutraceutical supplement pill (CNSP) stimulates naïve adult telomerase positive stem cells in-situ to reverse signs and symptoms in multiple health conditions. GSC Advan Res Rev. 2024; 20(02): 047-056 [201].
Therefore, endogenous adult telomerase positive stem cells and/or CNSP are both safe and effective at reversing the signs and symptoms in 46 health concerns in adult humans [Table 7, Table 8].
Part 8: Safety, responsibility, and Scientific Standards
Why introducing any stem cell category, safety is one of the first questions.
In its classical form, the Hippocratic Oath includes commitments to:
At a high level, what safety questions should scientists and physicians ask about aTPSCs?
Table 9. Safety Questions to be Asked.
|
Questions ?? |
Answers |
|
Do aTPSCs spontaneously differentiate |
No, they are under tight biological control |
|
Are aTPSCs tightly controlled |
Yes, they are under tight biological control |
|
How are aTPSCs controlled |
With biological agents: proliferation, progression, induction, and anti-differentiation |
|
What is their naïve default state |
Quiescent, hibernating, dormant cells |
|
Will aTPSCs form tumors |
None seen in 50+ years |
|
Will aTPSCs mutate |
Only when doubling times is less than cell cycle time |
|
Will aTPSCs form something other than what is wanted |
The body chooses to use activated aTPSCs as it sees fit. Life threating injuries are treated first, then other injuries are treated in reverse chronological order of appearance |
One concern with highly potent cells is tumor formation
Without going into disease-specific details, what should people understand about how aTPSCs differ from ESCs in this area, and I might add that iPSCs react the same as ESCs
Table 10. Comparison / Contrast of aTPSCs vs ESCs/iPSCs.
|
Characteristics |
aTPSCs |
ESCs / iPSCs |
|
Default state of naïve cells |
Quiescent, dormant cells |
Spontaneous differentiation |
|
Transplanted in naïve state |
Controlled by biological agents* |
Spontaneous differentiation into all cell types of the body |
|
End Result |
aTPSCs restore/regenerate the damaged and missing tissues |
Teratoma formation |
*aTPSCs are tightly controlled at each step of their repair process.
One major theme in regenerative medicine is whether cells come from the patient or a donor
Can you explain why using a patient on cells may matter?
Every somatic cell in the body contains cell surface self-recognition molecules for the body’s immune system to recognize whether a cell or any other entity (ECM, proteins, bacteria, viruses, fungi, mold, etc.) are self or non-self: hematopoietic cells express HLA-DR molecules, while other somatic cells express MHC Class-1 cell surface self-recognition molecules.
A progenitor stem cell, or GLSCs, EctoSCs, MesoSCs, and EndoSCs, from the same person exhibit the same self-recognition molecules and therefore is not destroyed by the immune system
At a simple level, why does the immune system care whether cells are self or non-self?
A non-self-cell (or any other entity: bacteria, viruses, parasites, fungi, mold, drugs, etc.) can pose a serious threat to the life of the individual. Therefore, the immune system (actually two systems) first destroys the invader with prejudice (innate immune system), chopping it up into small pieces to remember its characteristics (adaptive immune system) in case it ever appears in the future.
Can you explain GvHD is plain English?
GvHD is the body’s response to a foreign invader
What does it teach us about the limits of using cells from a different person?
If we use cells from a different individual, the self-recognition molecules need to either match the patient’s self-recognition molecules to prevent GvHD:
Or do not express self-recognition molecules in the undifferentiated state
What is the difference between a scientific discovery, an early clinical observation, and an accepted medical treatment?
Scientific Discovery
Is first to see an event, such as telomerase positive stem cells in regenerating adult salamander limbs
Early Clinical Observation
IRB approved clinical application for compassionate use and/or right to try for treatment to a few patients to determine
Accepted Medical Treatment
Why is the distinction important?
Prevents adverse harm to the patients
Before any new regenerative approach is widely accepted, what kind of evidence does the medical community need to see?
What should physicians be careful about when they hear exciting claims in regenerative medicine?
Part 9. The Big Picture of Regenerative Medicine
The videos introduce the idea of moving from an outside-in model to an inside-out model.
Do you agree with that framing?
Yes, I do, in two respects:
How would you explain it?
Is the future of regenerative medicine learning how to work with repair systems the body already has.
- Yes, to have a chance at successful regeneration requires an understanding of the model system (human body) which one is working with. The body has perfected repair / regeneration over multiple millenniums.
Do you think modern medicine has underestimated the body’s built-in capacity for repair?
- Yes, until recently, pre-1950’s (initiation of bone marrow transplants) the body was not thought to be able to repair itself.
- Scar tissue formation was considered normal healing
- Physicians did everything in their power to stimulate scar tissue formation
If the field begins to take aTPSCs seriously, how could that change the way researchers think about regenerative medicine?
Put your thinking caps on. Imagine what would be possible if you had access to a particular type of cell that was a universal donor and could be used for anyone, it is immunoprotected (invisible to immune system), it could proliferate to mass quantities (10^690+ doublings), could be changed into any cell type using specific inductive agents, it could be implanted as newborn cells in any aged individual. Basically, any living thing is possible.
The only limits to the capabilities of the aTPSCs are your imagination.
Could aTPSCs change the framework from “What cells can we add to the body?” to “What repair systems already exist in the body”
Short answer – YES
Can you introduce the idea that different conditions may require different cell populations?
Analogies to building a house: you built a two-story brick house in “tornado alley”, two years later a tornado completely destroys your house, the following is a potential sequence of events needed to restore your house to its original configuration.
You need to subcontract with different people to do specific jobs
Same with stem cells – you need specific cells to do specific jobs. What is unique about aTPSCs, especially TSCs and PSCs is that they can provide whatever cells are needed.
This series introduces the aTPSCs, their capabilities, how to isolate and propagate the cells, and their use in IRB-approved compassionate use / right to try human clinical trials.
Part 10. Closing Reflections
To reiterate Table 4. aTPSCs, especially TSCS, fit all the wish list criteria for the “Holy Grail” for regenerative medicine, and answer all the HYPE associated with the Holy Grail.
Table 4. Holy Grail Wish List (completed).
|
Attributes |
TSCs |
ESCs |
MSCs |
|
Telomerase Positive |
YES |
Yes |
No |
|
Unlimited proliferation Potential |
YES
|
Yes |
No |
|
Present throughout lifespan of individual |
YES |
NA |
Decrease with increasing age |
|
Absent Self-Recognition Molecules |
YES |
No |
No |
|
Invisible to Immune System |
YES |
No |
No |
|
Will form any somatic cell type |
YES |
Yes |
No, fat, cartilage, bone |
|
Does NOT spontaneously differentiate |
YES |
No |
Yes |
|
Pre-differentiation is NOT needed |
YES |
No |
Yes |
|
Will NOT form teratomas |
YES |
No |
Yes |
|
Function controlled by biological agents |
YES |
No |
Yes |
|
Homing receptors for damaged cells |
YES |
?? |
Yes |
|
Naïve state forms what is lost or damaged |
YES |
?? |
Yes |
|
Does NOT overgrow existing cells/tissues |
YES |
?? |
Yes |
|
Exosome Production |
YES |
Yes |
Yes |
|
Currently, can be propagated to large numbers without mutations |
YES |
No |
No |
|
Universal stem cell transplant |
YES |
No |
No |
|
Days of shelf-life at 4oC |
40 |
?? |
?? |
|
Cryopreserved |
-80oC |
-196 oC |
-196 oC |
|
Recovery Viability |
99% |
?? |
95% |
|
Can be Freeze Dried |
NYD |
No |
No |
|
Restoration Viability |
NYD |
No |
No |
|
Can withstand -196 oC to +200 oC |
NYD |
NYD |
NYD |
|
Bio-printed into 3D Constructs for transplant |
NYD |
NYD |
NYD
|
|
Additional Aspects |
|
|
|
|
Trypan Blue Staining |
YES |
No |
No |
|
Size, microns |
0.1-2 |
15-30 |
50-100 |
|
Traverse Blood-Brain Barrier |
YES |
No |
No |
|
Enter Thebesian System of Heart |
YES |
No |
No |
|
Heal heart from inside outward |
YES |
No |
No |
|
Replace all cells in damaged brains |
YES |
No |
?? |
|
Allogeneic cells can add new immune system without bone marrow ablation |
YES |
No |
?? |
|
Restore eyesight in dry macular degeneration |
YES |
No |
?? |
|
Reduce pain and increase ambulation in osteoarthritis |
YES |
?? |
?? |
CONCLUSION
The endogenous adult telomeres positive stem cells (aTPSCs), especially the totipotent stem cells (TSCS), fit all the wish list criteria for the “Holy Grail” for regenerative medicine, and can perform all the HYPE associated with the Holy Grail.
ACKNOWLEDGEMENTS
The authors wish to thank all the collaborators (co-authors on all papers) of Dr. Young for their input for these studies. Funding for these studies was provided by Rubye Ryle Smith Charitable Trust, MED Cen Foundation, and Dragonfly Foundation for Research and Development.
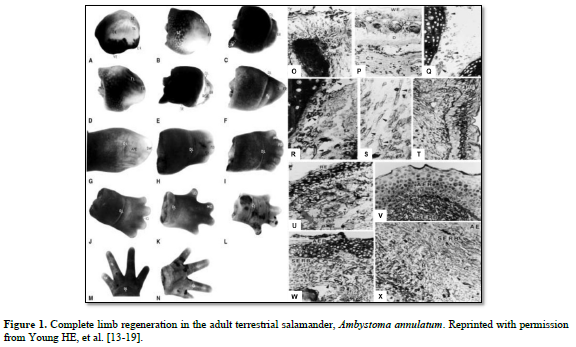
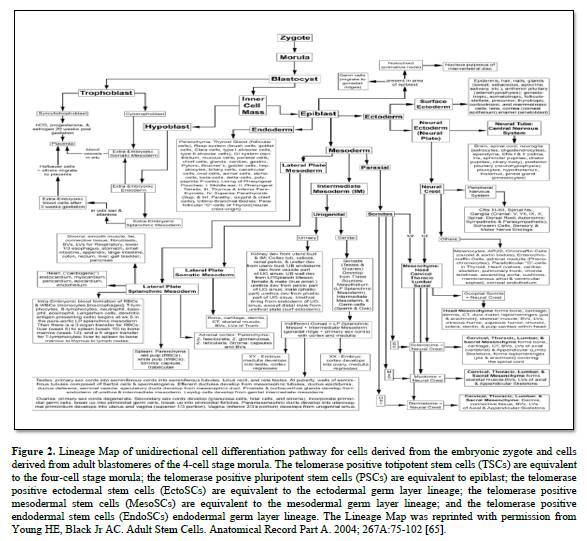
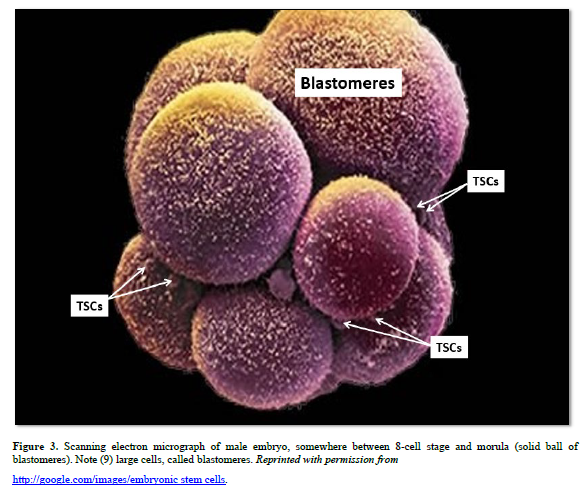
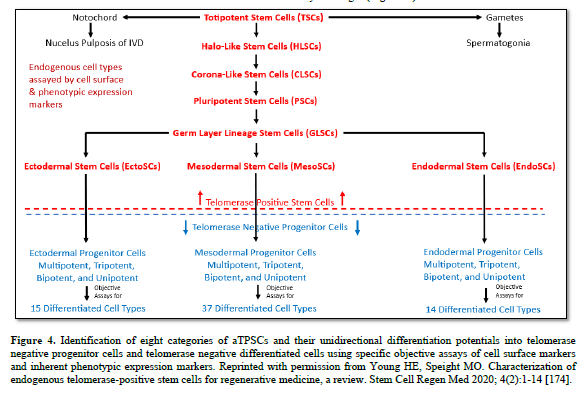
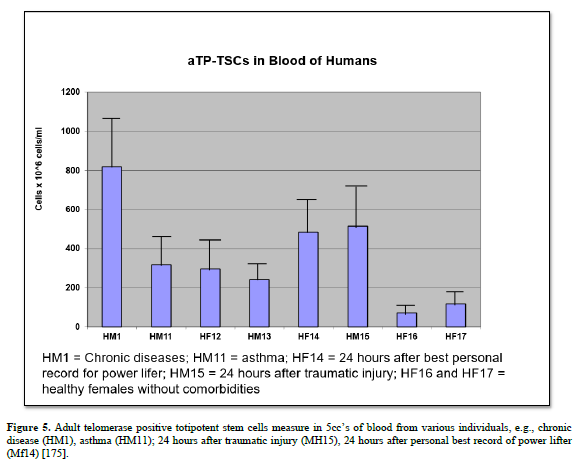
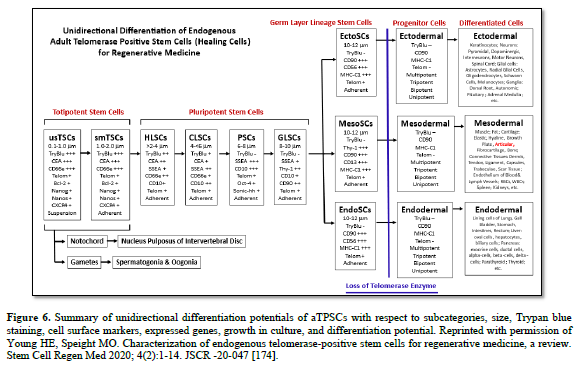

1. Farber J. An experimental analysis of regional organization in the regenerate forelimb of the axolotl (Ambystoma mexicanum). Arch Biol. 1959; 71:1-72.
2. Hay ED. Electron microscopic observations of muscle dedifferentiation in regenerating Amblystoma limbs. Dev Biol. 1959;1(6):555–585.
3. Gross J, Lapiere CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci U S A. 1962 Jun;48(6):1014–1022.
4. Thornton CS. Amphibian limb regeneration. In: Advances in Morphogenesis, Brachet J, King TJV eds. Academic Press, New York. 1968; 7: 205-249.
5. Dresden MH, Gross J. The collagenolytic enzyme of the regenerating limb of the newt Triturus viridescens. Dev Biol. 1970 May;22(1):129–157
6. Spotilla JR, Beumer RJ. The breeding habits of the ringed salamander, Ambystoma annulatum (Cope), in northwest Arkansas. Am Mid Natl. 1970; 84:77-89.
7. Toole BP, Gross J. The extracellular matrix of the regenerating newt limb: synthesis and removal of hyaluronate prior to differentiation. Dev Biol. 1971 May;25(1):57–77.
8. Scadding SR. Phylogenetic distribution of limb regeneration capacity in adult amphibia. J Exp Zool. 1972; 202:57-68.
9. Prichette WH, Dent JH. The role of size in the rate of limb regeneration in the adult newt. Growth. 1972; 36: 275-289.
10. Schauble MK. Seasonal variation of the newt forelimb regeneration under controlled environmental conditions. J Exp Zool. 1972; 181: 281-286.
11. Iten LE, Bryant. Forelimb regeneration from different levels of amputation in the newt, Notophthalamus viridescens: length, rate, stage. Wilhelm Roux Archiv. 1973; 173: 77-89.
12. Tank PW, Carlson BM, Connelly TG. A staging system for forelimb regeneration in the axolotl, Ambystoma mexicanum. J Morph. 1976; 150: 117-128.
13. Young HE. Limb Regeneration in the Adult Salamander, Ambystoma annulatum Cope 1889 (Amphibia: Ambystomatidae). University of Arkansas Library Press, copyright -1977;
14. Young HE. Epidermal ridge formation during limb regeneration in the adult salamander, Ambystoma annulatum. Proceedings of the Arkansas Academy of Science. 1977; 31:107-109;
15. Young HE, Bailey CF, Dalley BK. Environmental conditions prerequisite for complete limb regeneration in the postmetamorphic adult land-phase salamander, Ambystoma. Anatomical Record. 1983; 206:289-294;
16. Young HE, Bailey CF, Dalley BK. Gross morphological analysis of limb regeneration in postmetamorphic adult Ambystoma. Anatomical Record. 1983; 206:295-306;
17. Young HE. A Temporal Examination of Glycoconjugates During the Initiation Phase of Limb Regeneration in Adult Ambystoma. Texas Tech University Library Press, copyright 1983 and APPENDIX A Glycoconjugate Histochemistry Protocols, pp. 104-138.
18. Young HE, Dalley BK, Markwald RR. Identification of hyaluronate within peripheral nervous tissue matrices during limb regeneration. Edited by Coates, P.W., Markwald, R.R., Kenny, A.D., Alan R. Liss, Inc., New York. In: Developing and Regenerating Vertebrate Nervous Systems, Neurology and Neurobiology. 1983; 6:175-183;
19. Young HE, Bailey CF, Markwald RR, Dalley BK. Histological analysis of limb regeneration in postmetamorphic adult Ambystoma. Anatomical Record. 1985; 212:183-194.
20. Young HE, Speight MO. Allogeneic and autologous telomerase-positive stem cells as a potential treatment for systemic lupus erythematosus. Stem Cells Regen Med. 2020; 4(2):1-9.
21. Young HE, Young VE, Caplan AI. Comparison of fixatives for maximal retention of glycoconjugates for autoradiography, including use of sodium sulfate to release unincorporated radiolabeled [35S] sulfate. Journal of Histochemistry and Cytochemistry. 1989; 37:223-228.
22. Young HE, Carrino DA, Caplan AI. Histochemical analysis of newly synthesized and resident sulfated glycosaminoglycans during musculogenesis in the embryonic chick leg. Journal of Morphology. 1989; 201:85-103.
23. Young HE, Carrino DA, Caplan AI. Changes in synthesis of sulfated glycoconjugates during muscle development, maturation, and aging in embryonic to senescent CBF-1 mouse. Mechanisms of Ageing and Development. 1990; 53:179-193.
24. Young HE, Sippel J, Putnam LS, Lucas PA, Morrison DC. Enzyme-linked immuno-culture assay. Journal of Tissue Culture Methods. 1992; 14:31-36.
25. Young HE, Ceballos EM, Smith JC, Lucas PA, Morrison DC. Isolation of chick myosatellite and pluripotent stem cells. Journal of Tissue Culture Methods. 1992; 14:85-92.
26. Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991; 9:641-650.
27. Young HE, Ceballos EM, Smith JC, Mancini ML, Wright RP, Ragan BL, Bushell I, Lucas PA. Pluripotent mesenchymal stem cells reside within avian connective tissue matrices. In Vitro Cellular & Developmental Biology. 1993; 29A:723-736.
28. Rogers JJ, Adkison LR, Black AC Jr, Lucas PA, Young HE. Differentiation factors induce expression of muscle, fat, cartilage, and bone in a clone of mouse pluripotent mesenchymal stem cells. The American Surgeon. 1995; 61(3):231-236.
29. Young HE, Mancini ML, Wright RP, Smith JC, Black AC Jr, Reagan CR, Lucas PA. Pluripotent mesenchymal stem cells reside within the connective tissues of many organs. Developmental Dynamics. 1995; 202:137-144.
30. Young HE, Speight MO. Characterization of endogenous telomerase-positive stem cells for regenerative medicine, a review. Stem Cell Regen Med 2020; 4(2):1-14.
31. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999 Apr 2;284(5411):143–147.
32. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019 Dec 2; 4:22.
33. Gage FH. Mammalian neural stem cells. Science. 2000 Feb 25; 287(5457): 1433–1438.
34. Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. 1997; 8(6): 389–404.
35. Temple S. The development of neural stem cells. Nature. 2001; 414: 112–117.
36. Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005; 6(10): 777–788.
37. Ming G-L, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687–702
38. Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009; 32: 149–184.
39. Urbán N, Guillemot F. Neural stem cells: generating and regenerating the brain. Neuron. 2014; 83(1): 18–36.
40. Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U S A. 1992 Apr 1; 89(7): 2804–2808.
41. Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008 Nov 1; 112(9): 3543–3553.
42. Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010; 2(6): 640–653.
43. Kent DG, Copley MR, Benz C, Wöhrer S, Dykstra BJ, Ma E, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self renewal potential.
Blood. 2009 Jun 18; 113(25): 6342–6350.
44. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005 Jul 1; 121(7): 1109–1121.
45. Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018 Jan 18; 553(7689):418–426.
46. Wilkinson AC, Yamazaki S, Nakauchi H. Hematopoietic stem cells and their roles in tissue regeneration. Front Med (Lausanne). 2019; 6: 1–18.
47. Michalopoulos GK, Khan Z. Hepatic stem cells: in search of. Stem Cells. 2005; 23(6): 699 704.
48. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006; 43(2 Suppl 1): S45 53.
49. Turner R, Lozoya O, Wang Y, et al. Human hepatic stem cell and maturational liver lineage biology. Hepatology. 2011; 53(3): 1035 1045.
50. Petersen BE, Grossbard B, Goff JP, et al. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci U S A. 2003; 100(Suppl 1): 11881 11888.
51. Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003; 120(1): 117 130.
52. Tsamandas AC, Syrokosta I, Zolota V, et al. Oval cells in the liver: a putative stem cell compartment in chronic hepatitis and cirrhosis. Ann Gastroenterol. 2006; 19(3): 230 236.
53. Smukler SR, Arntfield ME, Razavi R, et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell. 2011; 8(3): 281 293.
54. Seaberg RM, Smukler SR, Kieffer TJ, et al. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Stem Cells. 2004; 22(7): 1070 1083.
55. Xu X, D’Hoker J, Stangé G, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008; 132(2): 197 207.
56. Inada A, Nienaber C, Katsuta H, et al. Carbonic anhydrase II–positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A. 2008; 105(50): 19915 19919.
57. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta cells are formed by self duplication rather than stem cell differentiation. Nature. 2004; 429(6987): 41 46.
58. Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. Basal cells are a stem cell population in the normal adult mouse trachea. Proc Natl Acad Sci U S A. 2004; 101(7): 2646 2651.
59. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005; 121(6): 823 835.
60. Giangreco A, Reynolds SD, Stripp BR. Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchioalveolar duct junction. Am J Pathol. 2002; 161(1): 173 182.
61. Engelhardt JF. Stem cells in the lung. In: Stripp BR, Reynolds SD, editors. Stem Cells in the Lung. New York: Marcel Dekker; 2005. p. 1 25.
62. Kajstura J, Rota M, Hall SR, Hosoda T, D’Amario D, Sanada F, et al. Identification of novel resident pulmonary stem cells: form and function of lung stem cells. Stem Cells. 2005; 23(8): 1073 1085.
63. Kotton DN, Fine A. Lung stem cells: new paradigms. Thorax. 2005; 60(3): 189 190.
64. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 6: 282(5391):1145-7.
65. Young HE, Black Jr AC. Adult Stem Cells. Anatomical Record Part A. 2004; 267A:75-102.
66. Nugent CI, Lundblad V. The telomerase reverse transcriptase: structure and function. Microbiol Mol Biol Rev. 2002;66(3):407–425.
67. Schmidt JC, Cech TR. Telomerase: mechanism of telomere synthesis. Annu Rev Biochem. 2012; 81:243–264.
68. Shammas MA. Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care. 2011;14(1):28–34.
69. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019;20(5):299–309
70. Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988; 336(6200):688–690.
71. Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988; 336(6200):688–690.
72. Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998; 12(13):2048–2060.
73. Ohtsuka S, Dalton S. Regulation of embryonic stem cell self-renewal and pluripotency by the LIF signaling pathway. J Biochem. 2008; 144(6):713–718.
74. Onishi K, Zandstra PW. LIF signaling in stem cells and development. Development. 2015; 142(13):2230–2236.
75. Onishi K, Zandstra PW. LIF signaling in stem cells and development. Development. 2015; 142(13):2230–2236.
76. Humphrey RK, Beattie GM, Lopez AD, Bucay N, King CC, Firpo MT, et al. Maintenance of pluripotency in human embryonic stem cells is not dependent on LIF/STAT3 signaling. Stem Cells. 2004; 22(5):770–778.
77. Tang C, Lee AS, Volkmer JP, Sahoo D, Nag D, Mosley AR, et al. An antibody against SSEA 5 glycan on human pluripotent stem cells enables removal of teratoma forming cells. Nat Biotechnol. 2011; 29(9): 829 834.
78. Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007; 25(9): 1015 1024.
79. Kehat I, Kenyagin Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into cardiomyocytes, exhibit functional properties of heart cells, and integrate into the rat heart. J Clin Invest. 2001; 108(3): 407 414.
80. Nussbaum J, Minami E, Laflamme MA, Virag JA, Ware CB, Masino A, et al. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J. 2007; 21(7): 1345 1357.
81. Lee HJ, Lim IJ, Park SW, Kim YH, Nam HY, Jeon YJ, et al. Human embryonic stem cell derived neural precursors prevent teratoma formation by differentiation and withdrawal of pluripotent cells. Stem Cells Dev. 2012; 21(4): 594 602.
82. Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013; 19(8): 998 1004.
83. Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009; 2(3): 198 210.
84. Moore KL, Persaud TVN, Torchia MG. The Developing Human: Clinically Oriented Embryology. 11th ed. Philadelphia: Elsevier; 2020. p. 25 56.
85. Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology. 6th ed. Philadelphia: Elsevier; 2021. p. 35 70.
86. Moore KL, Dalley AF, Agur AMR. Clinically Oriented Anatomy. 8th ed. Philadelphia: Wolters Kluwer; 2018.
87. Lee AS, Xu D, Plews JR, Nguyen PK, Nag D, Lyons JK, et al. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Transplant. 2009; 18(12): 1249 1257.
88. Gertow K, Hirst CE, Stanley EG, Elefanty AG. Standardization of the teratoma assay for analysis of pluripotency of human ES cells and iPS cells. PLoS One. 2012; 7(9): e45532.
89. Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009; 2(3): 198 210.
90. Mueller Klieser W, Tonn T, Heidenreich O, et al. Teratomas derived from embryonic stem cells as models for tumor biology. In: Schatten H, editor. Teratomas Derived from Embryonic Stem Cells as Models for Tumorigenesis. Rijeka: IntechOpen; 2011. p. 1 23.
91. Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro and teratoma formation in vivo. Nat Biotechnol. 2000; 18(4): 399 404
92. Lucas PA, Calcutt AF, Southerland SS, Wilson JA, Harvey RL, Warejcka D, Young HE. A population of cells resident within embryonic and newborn rat skeletal muscle is capable of differentiating into multiple mesodermal phenotypes. Wound Repair and Regeneration. 1995; 3:449-460;
93. Warejcka DJ, Harvey R, Taylor BJ, Young HE, Lucas PA. A population of cells isolated from rat heart capable of differentiating into several mesodermal phenotypes. J Surg Res. 1996; 62:233-242;
94. Lucas PA, Warejcka DJ, Zhang L-M, Newman WH, Young HE. Effect of rat mesenchymal stem cells on the development of abdominal adhesions after surgery. J Surg Res. 1996; 62:229-232;
95. Lucas PA, Warejcka DJ, Young HE, Lee BY. Formation of abdominal adhesions is inhibited by antibodies to transforming growth factor-beta1. J Surg Res. 1996; 65:135-138;
96. Dixon K, Murphy RW, Southerland SS, Young HE, Dalton ML, Lucas PA. Recombinant human bone morphogenetic proteins-2 and 4 (rhBMP-2 and rhBMP-4) induce several mesenchymal phenotypes in culture. Wound Repair and Regeneration. 1996; 4:374-380;
97. Young HE, Wright RP, Mancini ML, Lucas PA, Reagan CR, Black AC Jr. Bioactive factors affect proliferation and phenotypic expression in pluripotent and progenitor mesenchymal stem cells. Wound Repair and Regeneration. 1998; 6(1):65-75;
98. Young HE, Rogers JJ, Adkison LR, Lucas PA, Black AC Jr. Muscle morphogenetic protein induces myogenic gene expression in Swiss-3T3 cells. Wound Rep Reg. 1998; 6(6):543- 554;
99. Young HE, Steele T, Bray RA, Detmer K, Blake LW, Lucas PA, Black AC Jr. Human progenitor and pluripotent cells display cell surface cluster differentiation markers CD10, CD13, CD56, CD90 and MHC Class-I. Proc Soc Exp Biol Med. 1999; 221:63-71;
100. Young HE. Pluripotent stem cells. Edited by M.A. Brown and S. Neufield, Cambridge Healthtech Institute Press, Newton Upper Falls, MA. In: Second Annual Symposium on Tissue Engineering / Regenerative Healing / Stem Cell Biology. 1999; 469-530;
101. Young HE. Stem cells and tissue engineering. In: Gene Therapy in Orthopaedic and Sports Medicine, J. Huard and F.H. Fu, eds., Springer-Verlag New York, Inc., Chap. 9, pg. 143-173, 2000;
102. Young HE, Duplaa C, Young TM, Floyd JA, Reeves ML, Davis KH, Mancini GJ, Eaton ME, Hill JD, Thomas K, Austin T, Edwards C, Cuzzourt J, Parikh A, Groom J, Hudson J, Black AC Jr. Clonogenic analysis reveals reserve stem cells in postnatal mammals. I. Pluripotent mesenchymal stem cells. Anat. Rec. 263:350-360, 2001.
103. Young HE, Steele T, Bray RA, Hudson J, Floyd JA, Hawkins K, Thomas K, Austin T, Edwards C, Cuzzourt J, Duenzl M, Lucas PA, Black AC Jr. Human reserve pluripotent mesenchymal stem cells are present in the connective tissues of skeletal muscle and dermis derived from fetal, adult, and geriatric donors. Anat. Rec. 264:51-62, 2001.
104. Romero-Ramos M, Vourc’h P, Young HE, Lucas PA, Wu Y, Chivatakarn O, Zaman R, Dunkelman N, El-Kalay MA, Chesselet M-F. Neuronal differentiation of stem cells isolated from adult muscle. J Neurosci Res 69:894-907, 2002.
105. Young HE. Existence of reserve quiescent stem cells in adults, from amphibians to humans. Curr Top Microbiol Immunol. 280:71-109, 2004.
106. Young HE, Duplaa C, Romero-Ramos M, Chesselet M-F, Vourc’h P, Yost MJ, Ericson K, Terracio L, Asahara T, Masuda H, Tamura-Ninomiya S, Detmer K, Bray RA, Steele TA, Hixson D, El-Kalay M, Tobin BW, Russ RD, Horst MN, Floyd JA, Henson NL, Hawkins KC, Groom J, Parikh A, Blake L, Bland LJ, Thompson AJ, Kirincich A, Moreau C, Hudson J, Bowyer III FP, Lin TJ, Black Jr AC. Adult reserve stem cells and their potential for tissue engineering. Cell Biochem Biophys, 40(1):1-80, 2004.
107. Young HE, Duplaa C, Yost MJ, Henson NL, Floyd JA, Detmer K, Thompson AJ, Powell SW, Gamblin TC, Kizziah K, Holland BH, Boev A, Van de Water JM, Godbee DC, S. Jackson, M. Rimando, Edwards CR, Wu E, Cawley C, Edwards PD, Macgregor A, Bozof R, Thompson TM, Petro Jr GJ, Shelton HM, McCampbell BL, Mills JC, Flynt FL, Steele TA, Kearney M, Kirincich-Greathead A, Hardy W, Young PR, Amin AV, Williams RS, Horton MM, McGuinn S, Hawkins KC, Ericson K, Terracio L, Moreau C, Hixson D, Tobin BW, Hudson J, Bowyer III FP, Black Jr AC. Clonogenic analysis reveals reserve stem cells in postnatal mammals. II. Pluripotent epiblastic-like stem cells. Anat. Rec. 277A:178-203, 2004.
108. Vourc'h P, Romero-Ramos M, Chivatakarn O, Young HE, Lucas PA, El-Kalay M, Chesselet M-F. Isolation and characterization of cells with neurogenic potential from adult skeletal muscle. Biochemical and Biophysical Research Communications 317:893-901, 2004.
109. Seruya M, Shah A, Pedrotty D, du Laney T, Melgiri R, McKee JA, Young HE, Niklason LE. Clonal Population of adult stem cells: life span and differentiation potential. Cell Transplant 13:93-101, 2004
110. Young HE, Black AC Jr. Differentiation potential of adult stem cells. In: Contemporary Endocrinology: Stem Cells in Endocrinology, L.B. Lester, ed., The Humana Press Inc., Totowa, NJ. Chap. 4, p. 67-92, 2005b.
111. Vourc’h P, Lacar B, Mignon L, Lucas PA, Young HE, Chesselet MF. Effect of neurturin on multipotent cells isolated from the adult skeletal muscle. Biochem Biophys Res Commun 332:215-223, 2005.
112. Henson NL, Heaton ML, Holland BH, Hawkins KC, Rawlings B, Eanes E, Bozof R, Powell S, Grau R, Fortney J, Peebles B, Kumar D, Yoon JI, Godby K, Collins JA, Sood R, Bowyer 3rd FP, Black Jr AC, Young HE. Karyotypic analysis of adult pluripotent stem cells. Histology and Histopathology, 20: 769-784, 2005.
113. Mignon L, Vourc'h P, Romero-Ramos M, Osztermann P, Young HE, Lucas PA, Chesselet MF. Transplantation of multipotent cells extracted from adult skeletal muscles into the adult subventricular zone of adult rats. J Comp Neurol 491:96-108, 2005.
114. Young HE, Duplaa C, Katz R, Thompson T, Hawkins KC, Boev AN, Henson NL, Heaton M, Sood R, Ashley D, Stout C, Morgan JH, Uchakin PN, Rimando M, Long GF, Thomas C, Yoon JI, Park JE, Hunt DJ, Walsh NM, Davis JC, Lightner JE, Hutchings AM, Murphy ML, Boswell E, McAbee JA, Gray BM, Piskurich J, Blake L, Collins JA, Moreau C, Hixson D, Bowyer FP, Black AC Jr. Adult-derived stem cells and their potential for tissue repair and molecular medicine. J Cell Molec Med 9:753-769, 2005.
115. Young HE, Black AC Jr. Adult-derived stem cells. Minerva Biotechnologica Cancer Gene Mechanisms and Gene Therapy Reviews 17:55-63, 2005.
116. Stout CL, Ashley DW, Morgan III JH, Long GF; Collins JA, Limnios JI, Lochner F, McCommon G, Hixson D, Black Jr AC, Young HE. Primitive stem cells reside in adult swine skeletal muscle and are mobilized into the peripheral blood following trauma. American Surgeon 73 (11):1106-1110, 2007.
117. Stout CL, McKenzie J, Long G, Henson N, Hawkins KC, Ashley DW, Collins J, Hixson D, Black Jr AC, Young HE. Discovery of pluripotent and totipotent stem cells in the heart of the adult rat. Amer Surg 73: S63, 2007.
118. Vourc’h P, Mignon L, Lucas PA, Young HE, Chesselet MF. Cells isolated from adult skeletal muscle express markers of differentiated neurons after transplantation into the adult hippocampus. (In press).
119. Young HE and Black Jr AC. Naturally occurring adult pluripotent stem cells. In: Stem Cells: From Biology to Therapy, Advances in Molecular Biology and Medicine. 1st Ed, R.A. Meyers, Ed, WILEY-BLACKWELL-VCH Verlag GmbH & Co. KGaA. Chap 3, pp. 63-93, 2013.
120. McCommon GW, Lochner F, Black Jr AC, Young HE. Primitive adult-derived stem cells are present in the blood of adult equines and can be increased in number with moderate exercise or ingestion of a cyanobacter, Aphanizomenon flos-aquae. Autocoids 2: 103, 2013,
121. Young HE, Henson NL, Black GF, Hawkins KC, Coleman JA, Black Jr AC. Location and characterization of totipotent stem cells and pluripotent stem cells in the skeletal muscle of the adult rat. J Stem Cell Res 1(1) 002: 1-17, 2017.
122. Young HE, Lochner F, Lochner D, Lochner D, McCommon G, Black AC Jr. Primitive Stem Cells in Adult Feline, Canine, Ovine, Caprine, Bovine, and Equine Peripheral Blood. J Stem Cell Res. 1(1) 004: 1-6, 2017.
123. Young HE, Lochner F, Lochner D, Lochner D, Black GF, Coleman JA, Young VE, McCommon G, Black Jr AC. Primitive stem cells in adult human peripheral blood. J Stem Cell Res. 1(2) 001:1-8, 2017.
124. Young HE, Henson NL, Black GF, Hawkins KC, Coleman JA, Black Jr AC. Stage-Specific Embryonic Antigen-4-Positive Cells and Carcinoembryonic Antigen Cell Adhesion Molecule-1-Positive Cells are Located in the Bone Marrow of the Adult Rat. J Stem Cell Res. 1(2) 001: 1-3, 2017.
125. Young HE, Limnios JI, Lochner F, McCommon G, Black GF, Coleman JA, Hawkins KC, Black Jr AC. Healing cells in the dermis and adipose tissue of the adult pig. J Stem Cell Res 2017; 1(2) 004:1-5.
126. Young HE, Black GF, Coleman JA, Hawkins KC, Williams S, Black Jr AC. Healing cells in the kidney of the adult rat. J Stem Cell Res 2017; 1(3) 001:1-4.
127. Young HE, Limnios JI, Lochner F, McCommon G, Black GF, Coleman JA, Hawkins KC, Black Jr AC. Healing cells in the dermis and adipose tissue of the adult pig. J Stem Cell Res 2017; 1(2) 004:1-5.
128. Young HE, Lochner F. Telomerase positive totipotent stem cells in the adult brain. I. cerebral cortex. Regen Med Biol Res 2021; 2(1):1-16.
129. Young HE, Lochner F. Endogenous adult telomerase positive stem cells increase in equine peripheral blood following exercise. J Reg Med Biol Res. 2024; 5(2):1-8.
130. Young HE. Carcinoembryonic antigen-cell adhesion molecule-1 and stage-specific embryonic antigen-4 are present in the reproductive organs of adult mammals. GSC Advanced Research and Reviews. 2025; 23(03): 149-157.
131. Young HE. Totipotent stem cells and pluripotent stem cells are present in the reproductive organs of an adult mammal. GSC Advanced Research and Reviews. 2025; 23(03): 158-180.
132. Yamanaka S. A fresh look at iPS cells. Cell. 2009;137(1):13 17.
133. Takahashi K, Narita M, Yokura M, Ichisaka T, Yamanaka S. Human induced pluripotent stem cells on autologous feeders. PLoS One. 2009; 4(12): e8067.
134. Ohnuki M, Takahashi K, Yamanaka S. Generation and characterization of human induced pluripotent stem cells. Curr Protoc Stem Cell Biol. 2009; Chapter 4: Unit 4A.2.
135. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012; 13(10): 693 704.
136. Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015; 350(6265): 1193 1198.
137. Lansdorp PM. Telomeres and telomerase: a simple end matter. Nat Med. 2005;11(10):1049 1054.
138. Schmidt JC, Cech TR. Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015; 29(11): 1095 1105.
139. Kordinas V, Tsirpanlis G, Imprialos K, Giannopoulou E, Doumas M. Telomere length, telomerase activity and atherosclerosis. Curr Med Chem. 2016; 23(12): 1280 1290.
140. Batista LFZ, Artandi SE. Telomere and telomerase dynamics in induced pluripotent stem cells. World J Stem Cells. 2013; 5(3): 116 123.
141. Ahmed MS, Ikram S, Bibi N, Mir A. Telomere biology in induced pluripotent stem cells: implications for aging and regenerative medicine. World J Stem Cells. 2016; 8(6): 182 194.
142. Jenkins YM, de Lange T. Telomerase and induced pluripotency. Cell Res. 2012; 22(4): 614 617.
143. Wang J, Xie LY, Allan S, Beach D, Hannon GJ. Myc activates telomerase. Genes Dev. 1998; 12(12): 1769 1774.
144. López de Si.lanes I, Graña O, De Bonis ML, Dominguez O, Pisano DG, Blasco MA. Identification of TERRA locus unveils a telomerase regulating role for telomeric RNAs in iPSC. Cell. 2014; 158(6): 1232 1245.
145. Tomov ML, Yamada S, Pace J, et al. iPSC culture: what every researcher needs to know. Tempo Biosci Technical Bulletin. 2025; 1: 1 12.
146. JangoCell Scientific Team. Detecting and preventing spontaneous differentiation in iPSC and MSC cultures. JangoCell Application Note. 2025; 1: 1 10.
147. Stoilova T, Hristova A, Kolev M, et al. Improving the differentiation potential of pluripotent stem cells by controlling spontaneous differentiation. Sci Rep. 2022; 12(1): 14321.
148. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Proc Natl Acad Sci U S A. 2009; 106(33): 12741 12746.
149. Gertow K, Hirst CE, Stanley EG, Elefanty AG. Teratoma formation: a tool for monitoring pluripotency in stem cell research. Curr Protoc Stem Cell Biol. 2015; 32: 4A.8.1 4A.8.23.
150. Ben David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011; 11(4): 268 277.
151. Young HE, Black AC. Pluripotent Stem Cells, Endogenous versus Reprogrammed, a Review. MOJ Orthop Rheumatol 1(4): 00019, 2014.
152. Young HE, Black GF, Coleman JA, Hawkins KC, Black Jr AC. Pulmonary diseases and adult healing cells: from bench top to bedside. J Stem Cell Res 2017; 1(2) 003:1-9.
153. Young HE, Hyer L, Black AC Jr, Robinson Jr JS. Treating Parkinson disease with adult stem cells. J Neurological Disorders, 2:107-109, 2013b.
154. Young HE, Hyer L, Black AC Jr, Robinson Jr JS. Adult stem cells: from bench-top to bedside. In: Tissue Regeneration: Where Nanostructure Meets Biology, 3DBiotech, North Brunswick, NJ Chap 1, pp 1-60, 2013a.
155. Young HE, Limnios IJ, Lochner F, McCommon G, Black GF, Coleman JA, Hawkins KC, Black Jr AC. Cardiovascular disease and adult healing cells: From bench top to bedside. J Stem Cell Res 2017; 1(3) 002:1-8.
156. Black Jr AC, Williams S, Young HE. From Bench Top to Bedside: Formation of Pulmonary Alveolar Epithelial Cells by Maintenance Cells and Healing Cells. J Stem Cell Res. 2017; 1(2) 002: 1-16.
157. Young HE, Speight MO. Local Anesthetics Can Affect the Efficacy of Telomerase-Positive Stem Cells. J Regen Med. Biol Res 2020; 1(1):1-9.
158. Young HE, Speight MO. Osteoarthritis Treated with Telomerase-Positive Adult Stem Cells in Animals and Humans. Stem Cells Regen Med. 2020; 4(2):1-11. JSCR-20-049.
159. Young HE, Speight MO. Informed consent guidelines for optimizing the use of telomerase-positive stem cells for regenerative medicine. J Regen Med Biol Res 2020; 1(1):1-20.
160. Young HE, Speight MO. Telomerase-positive stem cells as a potential treatment for idiopathic pulmonary fibrosis. Stem Cells Regen Med. 2020; 4(2):1-11.
161. Young HE, Speight MO. Criteria to Distinguish TSCs from Exosomes as Major Players in Regenerative Medicine. J Regen Med & Biol Res. 2020; 1(1):1-5. JRMBR-1(1)-005.
162. Young HE, Speight MO. Potential treatment of chronic obstructive pulmonary disease with allogeneic and autologous telomerase-positive stem cells. Stem Cells Regen Med. 2020; 4(3):1-11. JSCR-20-056.
163. Young HE, Speight MO. Allogeneic telomerase-positive stem cells as a treatment for celiac disease. Stem Cells Regen Med. 2020; 4(2):1-7 JSCR-20-050.
164. Young HE, Speight MO. Cardiovascular disease treated with telomerase-positive stem cells. Stem Cells Regen Med. 2020; 4(2):1-8 JSCR-20-051.
165. Young HE, Speight MO. Age-related macular degeneration treated with autologous telomerase-positive totipotent stem cells. Stem Cells Regen Med. 2020; 4(3):1-9. JSCR-20-055.
166. Young HE, Speight MO. Donor selection is a critical component using naïve endogenous adult stem cells for the treatment of chronic diseases and traumatic injuries. J Regen Med & Biol Res. 2020; 1(2):1-28. JRMBR-1(2)-009.
167. Young HE, Speight MO. Alzheimer’s disease treated with autologous and allogeneic telomerase-positive stem cells. Stem Cells & Regen Med. 2021; 5(1):1-17.
168. Young HE, Speight MO. Blunt force trauma-induced total bilateral vision impairment of 13 years duration treated with autologous telomerase positive stem cells. Stem Cells Regen Med. 2021; 5(1):1-22.
169. Young HE, Speight MO. Traumatic spinal cord injury treated with autologous telomerase-positive stem cells. Stem Cells Regen Med. 2021; 5(1):1-13.
170. Young HE, Speight MO. Chronic inflammatory demyelinating polyneuropathy treated with autologous telomerase-positive stem cells. Stem Cells Regen Med. 2021; 5(2):1-16.
171. Young HE, Speight MO. Treating Parkinson Disease with Autologous Telomerase-Positive Stem Cells, Update 2021. Stem Cells & Regenerative Medicine. 2021; 5(1): 1-13.
172. Young HE. Healing Cells: Use What the Almighty Created to Heal Thyself. American Journal of Medical and Clinical Research & Reviews. 2023; 2(7):1-19.
173. Young HE. A high throughput screening assay to quantify, visualize, and standardize biological activities: Enzyme-Linked Immuno-Culture Assay (ELICA). GSC Advanced Research and Reviews. 2025; 24(02): 091-114.
174. Young HE, Speight MO. Characterization of endogenous telomerase-positive stem cells for regenerative medicine, a review. Stem Cell Regen Med 2020; 4(2):1-14.
175. Young HE. Endogenous Adult Telomerase Positive Stem Cells (aTPSCs) and Combinatorial Nutraceutical Supplement Pill (CNSP), 50+ Years in the Making, 50+ Years of Discovery; In preparation.
176. Caplan AI. Mesenchymal stem cells: Time to change the name! Stem Cells Transl Med. 2017; 6(6): 1445–1451.
177. Caplan AI, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011; 9(1): 11–15
178. Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006 Aug 1; 98(5): 1076–1084.
179. Dominici M, Le Blanc K, Mueller I, Slaper Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. Cytotherapy. 2006;8(4):315 317.
180. Lv F J, Tuan RS, Cheung KMC, Leung VYL. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32(6):1408 1419.
181. Samsonraj RM, Raghunath M, Nurcombe V, Hui JH, van Wijnen AJ, Cool SM. Concise review: multifaceted characterization of human mesenchymal stem cells for use in regenerative medicine. Stem Cells Transl Med. 2017;6(12):2173 2185.
182. Kuci S, Kuci Z, Schäfer R, Bruderek K, Schardt A, Blaeschke F, et al. Clonal analysis of multipotent stromal cells reveals limited hierarchies. Stem Cells. 2018;36(12):1909 1924.
183. Phinney DG, Pittenger MF. Concise review: MSC derived exosomes for cell free therapy. Stem Cells. 2017;35(4):851 858.
184. Young HE, Speight MO, Black AC Jr. Functional Cells, Maintenance Cells, and Healing Cells. J Stem Cell Res. 1(1): 003: 1-4, 2017.
185. Zvereva MI, Shcherbakova DM, Donsta OA. Telomerase: structure, functions, and activity regulation. Biochemistry (Mosc.) 2010; 75: 1563-1583.
186. Young HE. Combinatorial Nutraceutical Supplement Pill (CNSP) Stimulates Naïve Adult Telomerase Positive Stem Cells In-Situ to Heal Cardiomyopathies. GSC Advanced Research and Reviews. 2024; 20(02), 047-056.
187. Young HE. Fresh Isolate Adult Telomerase Positive Stem Cells: An addition to Embryonic Stem Cells (ESCs), Induced Pluripotent Stem Cells (iPSCs), and/or Mesenchymal Stem Cells (MSCs) for Regenerative Medicine. GSC Advanced Research and Reviews. 2023; 16(1):066-081.
188. Young HE. Adult telomerase positive stem cells: isolation, plating, and propagation. GSC Advanced Research and Reviews. 2025; 25(02):407-440.
189. Young HE, Limnios JI, Lochner F, McCommon G, Cope LA, Black AC Jr. Pancreatic islet composites secrete insulin in response to a glucose challenge. J Stem Cell Res 1(1) 001: 1-12, 2017.
190. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37(3):614 636.
191. Kumar V, Abbas AK, Aster JC. Robbins Basic Pathology. 10th ed. Philadelphia: Elsevier; 2018.
192. Young HE. Adult telomerase positive stem cells: introduction and location. GSC Advanced Research and Reviews. 2025; 25(02): 296-331.
193. Young HE. Adult telomerase positive stem cells: isolation, plating, and propagation. GSC Advanced Research and Reviews. 2025; 25(02):407-440.
194. Young HE. Adult telomerase positive stem cells: differential cryopreservation, cell surface marker profiles, and cell sorting. MOJ Orthopedics & Regeneration. 2025; 17(5): 155-173.
195. Young HE. Adult telomerase positive stem cells: Cell-Specific Exosome-Conditioned Medium, Repetitive Single Cell Clonogenic Analysis, and Genomic Labeling. GSC Advanced Research and Reviews. 2025; 25(2): 480-504.
196. Young HE. Adult telomerase positive stem cells: Effects of Biological Agents on Single Cell Clones of Adult Cells. GSC Advanced Research and Reviews. 2025; 25(03): 028-047.
197. Young HE. Adult Telomerase Positive Stem Cells: Effects of Biological Agents on Genomically-Labeled Clones and Unlabeled Clones of Adult Cells. GSC Advanced Research and Reviews. 2025; 25(03): 127-145.
198. Young HE. Adult Telomerase Positive Stem Cells: Compare and Contrast Biobanking with Mesenchymal Stem Cells and Other Progenitor Cells. GSC Advanced Research and Reviews. 2025; 25(03): 218-226.
199. Young HE. Adult Telomerase Positive Stem Cells: Induced Proliferation of Precursor Cells by Platelet-Derived Growth Factor-BB. J Stem Cell Res. 2026; 7(1):1-15.
200. Young HE. Adult Telomerase Positive Stem Cells: Remain constant throughout lifespan of individual. J Stem Cell Res. 2026; 7(1):1-13.
201. Young HE. Combinatorial nutraceutical supplement pill (CNSP) stimulates naïve adult telomerase positive stem cells in-situ to reverse signs and symptoms in multiple health conditions. GSC Advan Res Rev. 2024; 20(02): 047-056.
No Files Found




Internationally Accepted
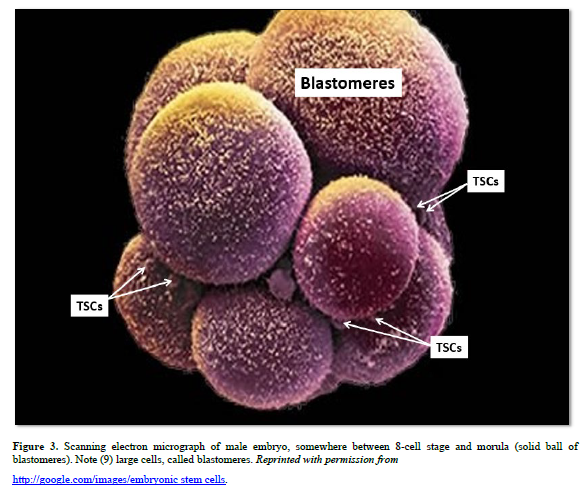
Share Your Publication :